 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
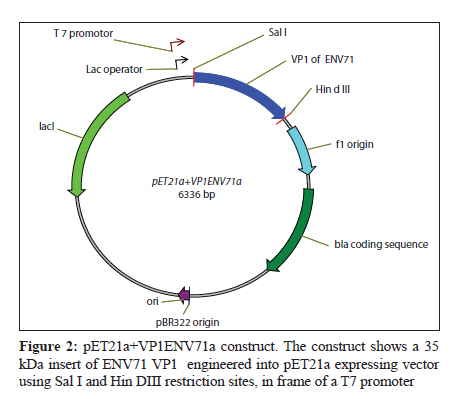
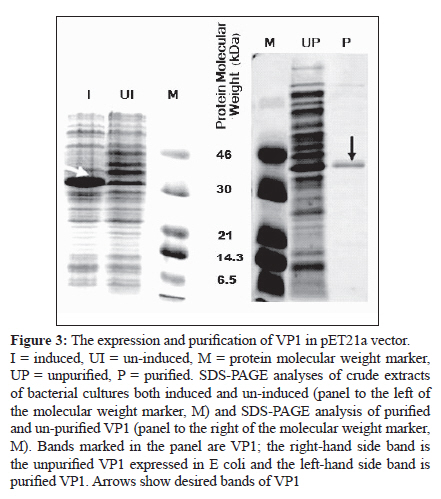
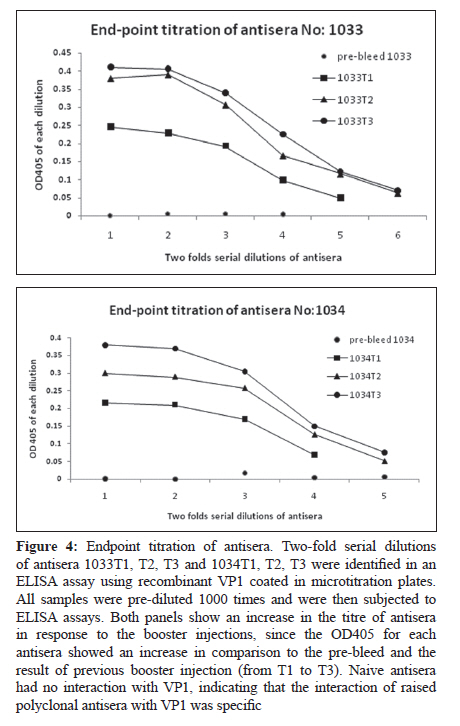
Indian Journal of Medical Microbiology, Vol. 28, No. 3, July-September, 2010, pp. 196-200 Original Article Development of polyclonal antisera to clone enterovirus 71 cellular receptor AB Sioofy-Khojine Department of Pathobiology, Room 304, Faculty of Veterinary Medicine, University of Tabriz, Tabriz - 5176614775, Fellow of Research Centre of Infectious Diseases & Tropical Medicine, Sina Gerenal Hospital, Tabriz, Iran Keywords: Enterovirus 71, polyclonal antisera, viral protein 1 Introduction Enterovirus 71 (ENV71) is a member of the family Picornaviridae, which was first reported from patients with neurologic disorders in California. [1] This virus is one of the classic causes of hand-foot-and-mouth disease. [2] The clinical symptoms of ENV71 infection in adults are diverse and have been reviewed by Chang et al. in 2004. [3] An emerging form of ENV71 - which causes a significant increase in the mortality (up to 6%) among infants and young children [4] - is of medical importance since it causes rapidly fatal neurogenic pulmonary oedema and heart failure, along with rhombencephalitis. [5] One of the possible reasons for the changes in the clinical features of the disease is that the receptor binding of the virus has changed. [6] There was no candidate for the virus binding at the time and hence we proposed to investigate receptor interaction of the virus. A monoclonal antibody that specifically blocks the virus binding to the target cells and/or infection is an essential requirement for receptor cloning purposes. Screening the available libraries containing antibodies to cell surface molecules that block ENV71 binding or infection did not generate fruitful results and; therefore, it was proposed that an anti-virus antibody be generated in order to perform ENV71 cellular receptor cloning study. Raising monoclonal antibodies is time consuming, whereas producing polyclonal antisera in rabbits is relatively rapid and generates antisera with wide reactivity (i.e., against more than one epitope on a target antigen). Ethical considerations prohibit the immunisation of experimental animals with a live virus. In order to raise polyclonal antisera (in rabbits), 50-100 ng of a target protein (intact 160S ENV71 particles) was required. To increase the possibility of obtaining an antiserum against the target protein it was recommended to immunise two rabbits in parallel (complying with the UK Home Office regulations regarding experimental animals). It was also necessary to repeat the injections three times to acquire a detectable level of immune response. Thus, it was calculated that a total amount of ~800 ng of viral protein was required for the immunisation. We estimated that this amount of protein would be provided by about 1.5 Χ 10 11 viral particles, assuming the molecular weight of the ENV71 to be 2.5 Χ 10 3 kDa (about the same molecular weight as PV3). Maximum yields of ENV71 in laboratory conditions was 5 Χ 10 7 TCID 50 ml−1 and, under an ideal purification condition, we estimated that the titre of the purified 160S particles recovered would be 5 Χ 10 5 TCID 50 ml−1 . From this, we calculated that at least 300 litres of infectious tissue culture supernatant of ENV71 would be required to recover 800 ng of viral capsid proteins. This was not a realistic approach because of the scale of the material to be concentrated and therefore we selected an alternative method, i.e., the expression of the immunogen protein of the virus in a eukaryotic system. Materials and Methods The cloning of the VP1-coding region of ENV71 A vector called pET21a (Novagen, Cambridge, UK) was selected to clone the VP1-coding region of ENV71. A consensus sequence of VP1 was generated using BioEdit package from the sequences published in the GenBank for VP1 sequence, [Figure - 1]. The consensus sequence was used to assess the presence of the unique restriction sites for cloning purposes. One pair of primers was designed to match either the 5′ or the 3′ ends of the VP1 consensus sequence of ENV71, harbouring the unique restriction sites of Hin dIII and Sal I, respectively, to allow unidirectional cloning for expression purposes. Individually published sequences were examined to ascertain that the restriction sites do not exist in the VP1-coding region of ENV71. Reverse transcriptase (RT) and polymerase chain reaction (PCR) were optimised and the VP1 region of ENV71 was amplified from a viral RNA preparation, using designated primers (data not shown). PCR products were purified using MicroSpin columns and were cloned into pET21a that carried the desired unique restriction sites (Hin dIII and Sal I) for the VP1 cloning. This plasmid carries an in-frame 6xHis Tag to allow the purification of the recombinant protein. The VP1-coding sequence was ligated into the vector, purified using MicroSpin columns, and propagated in the DH5α strain of E coli following transformation by electroporation. For further experiments, a large-scale alkaline lysis preparation of the clone was conducted using a Maxiprep kit (Qiagen, West Sussex, UK). Expression of VP1 in E coli strain BL21(DE3)pLys S The expression conditions of the pET21a-VP1 were optimised in E coli strain BL21(DE3)pLys S. The transformed bacteria were cultured in the presence of 0.2% w/v glucose in order to tightly repress the expression of the inserted gene prior to induction with isopropyl β-D-1-thiogalactopyranoside (IPTG). One colony was selected from a freshly streaked plate of pET21a-VP1 transfectants, cultured at 37°C, and then induced using IPTG and incubated for an additional 2 hours at 37°C to express the protein. The cells were harvested and then subjected to 15% SDS-PAGE analysis under reducing conditions and visualised after staining with Coomassie brilliant blue. Raising and quantification of rabbit polyclonal antisera to ENV71 VP1 Two rabbits (Elite New Zealand White) were housed for 7 days in the animal lab prior to pre-bleed. The pre-immune blood samples were collected from the rabbits and the serum was isolated from each sample. The naive sera were aliquoted in smaller quantities after inactivation of the complement activity and were stored at −20°C for future assays. The immunisation program was commenced 4 days after the pre-bleed (called day 0). On day 0, 100 ng of the purified recombinant VP1 was injected into each rabbit subcutaneously in a total volume of 400 μl containing 25% v/v complete Freund′s adjuvant (Sigma, Dorset, UK). On days 28, 42, and 56, three booster injections were administered subcutaneously to each rabbit, using the same amount of VP1 but in 25% v/v incomplete Freund′s adjuvant (Sigma). Ninety-six-well plates (Nunc) were coated with recombinant VP1 and were prepared for the assays. The antisera were applied to the wells in two-fold serial dilutions. The optical density was determined against the blank well (i.e., one containing no antigen and treated the same as the others) for each well at a wavelength of 405 nm (OD 405 ) after adding the substrate tetramethylbenzidine (TMB). The negative control wells were coated with the antigen to determine the reactivity of the secondary antibody (as a background binding). The results are adjusted to the baseline absorbance by deducting the values obtained from the negative control wells from the test results (see the results section). Results The presence of a correct size insert was confirmed by restriction digestion analysis of the alkaline lysis preparations (from single colonies of the transformants). The plasmid was successfully cloned and the construct was named pET21a+VP1ENV71a [Figure - 2]. IPTG-induced expression of ENV71 VP1 resulted in the production of significant levels of a protein of the expected size of 36.5 kDa in the induced samples containing pET21a+VP1ENV71a the white arrow pointing to VP1 in the lane designated ′I′ in [Figure - 3], left. Having established the expression method, large-scale expression of the VP1 was prepared. A freshly transfected BL21 strain of E coli (with pET21a+VP1ENV71a) was cultured on a small scale in the presence of 0.2% w/v glucose in selective media at 37°C for 24 hours. The overnight culture was then transferred into 500 mL Luria-Bertani (LB) broth containing the selective antibiotics but not glucose and was then incubated under the previously defined conditions. Cultures were then induced by IPTG for protein expression. The bacterial cells were harvested and lysed by one round of freezing and thawing and the VP1 tagged with 6xHis was purified using nickel columns. The presence of the correct size recombinant VP1 was shown in a denaturing reducing SDS-PAGE stained with Coomassie brilliant blue [Figure - 3], right. The animals were test-bled (10 mL) 7 days after the third booster injection (on day 63). Serum was separated from blood samples, inactivated, and stored at −20°C for future assays. The immune sera were titred against the purified recombinant VP1. Another booster injection was administered on day 70 to increase the titre of the polyclonal antisera and the animals were again test-bled on day 77. The last booster injection was delivered on day 84 and the rabbits were finally bled and euthanized on day 91. Serum samples obtained on day 63 (called 1033T1 and 1034T1), 77 (called 1033T2 and 1034T2), and 91(called 1033T3 and 1034T3). The serum samples were called so based on rabbit number followed by test-bleed number. The specific reactivity of the antisera was detected using recombinant VP1 and this reactivity was compared to other samples from the same rabbits. Purified recombinant VP1 was used in ELISA assays to determine the specific reactivity of the antisera. Initial ELISA assays showed that all antisera, except the pre-immune antisera, recognised the recombinant purified VP1 (data not shown). These experiments suggested that the antisera needed to be pre-diluted at least 1000 times to conduct a titration assay. The titre of the antisera for each rabbit showed an increase in response to the boost injections. The endpoint titration of 1033T1-3 and 1034T1-3 was performed using pre-diluted (1000 times) antisera in an ELISA assay as before. Since the sensitivity of the TMB for the identification of positive wells was not determined, the values below OD 405 of 0.05 were assumed as background. The end-point titre for the immune antisera were at about 1/64000 for 1033T2, 3, 1/32000 for 1034T2, 3, and 1/16000 for the rest of the immune antisera. ELISA assays also revealed that the antisera recognised the linear epitopes of the recombinant VP1. It was, however, not known if the antisera would recognise the native form of the VP1 present on the intact viral capsid or not. It was also shown that the immune response to the immunogen peptide was stronger in rabbit number 1033 than in 1034, as it was to the recombinant VP1 [Figure - 4]. The attempts to detect the ENV71-full particles immobilised on microtitre plates (in an ELISA test format) were not successful since it did not produce consistent results. This could be because of improper (and/or inadequate) immobilisation of the viral particles, or because the immobilised particles were blocked with the BSA present in the blocking agent. ELISA on intact enterovirus particles is poorly reproducible and it is an assay that is not routinely used in picornavirus laboratories. For this reason the reactivity of the antisera was tested against ENV71 particles. The infectivity of ENV71 infectious particles was blocked in the presence of both immune antisera, showing that the antisera recognise native epitopes on intact viral particles. This work was essentially in line with the results and recommendations of other works, such as the report by Malaviya[7] or the recent publication by Leenaars and Hendriksen. [8] Discussion A number of different methods have been employed to identify viral receptors/co-receptors for animal viruses. A variety of approaches, including genetic, immunological, co-purification, and screening methods, have been used in different studies to identify viral receptors. In some cases the receptor molecule was identified using a single method, while in others multiple approaches were required. Antibodies are efficient tools for viral receptor research. Some antibodies have been successfully used to identify viral receptors, for example the receptors of measles virus [9],[10] and Sindbis virus. [11] Transferring the gene has also been used extensively to identify viral receptors. The transfection of genes was shown to confer on non-permissive cells the ability to support virus binding and, possibly, infection. Cloning of the poliovirus receptor is a typical example of a genetic approach used to identify a viral receptor. [12] Viral receptor discovery has also benefited from a technique called virus overlay protein binding assay (VOPBA), which is also known as overlay blot assay. The cellular receptor for the minor group of human rhinoviruses was identified using an overlay assay and subsequently characterised as the low-density lipoprotein receptor. [13] A number of additional methods were also employed in some cases to further examine the specificity of the identified receptor. This study outlined the framework for a receptor study and produced the basic component of the receptor cloning experiment by producing a VP1-reactive antibody. Antigenic regions of picornaviruses have been previously studied by using synthetic peptides, peptide scanning, and neutralization escape mutants. In poliovirus type 1 and 3 (PV1 and PV3), coxsackie virus type A9 (CV-A9), human rhinovirus 14 (HRV14), and foot-and-mouth disease virus (FMDV), the major antigenic regions frequently contain amino acid sequences from the capsid protein VP1, whereas VP2 and VP3 are less often involved in the antigenic structure. [14],[15],[16],[17],[18] There was no reason to assume that this was not true in the case of ENV71 and hence the VP1 was selected for raising polyclonal antisera. The overall assessment of both antisera revealed reactivity against recombinant VP1 as well as against native ENV71 VP1. It was shown that serum samples from patients infected with ENV71 recognise linear epitopes of a recombinant VP1, [19] indicating that these epitopes are immunogenic when a productive infection is progressing in vivo. The finding of our study is also in line with these reports. Cloning, expression, and purification of the recombinant VP1 were successful. Immunising two rabbits generated specific polyclonal antisera that recognise viral capsid protein VP1 in ELISA assays [Figure - 4] and neutralise infectious viral particles (data not shown) and could therefore be used for receptor cloning purposes using immuno-focal screening methods. Acknowledgments I am grateful to Professor D. J. Evans who provided the supervision for the project and Dr. IG. Goodfellow and Dr. DT. Williams for their instructive advice and critical reading of this manuscript. This work was funded by the Ministry of Science, Research and Technology of Iran through The University of Tabriz, Iran. References
Copyright 2010 - Indian Journal of Medical Microbiology The following images related to this document are available:Photo images[mb10063f1.jpg] [mb10063f4.jpg] [mb10063f2.jpg] [mb10063f3.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}
{kind=link}