 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
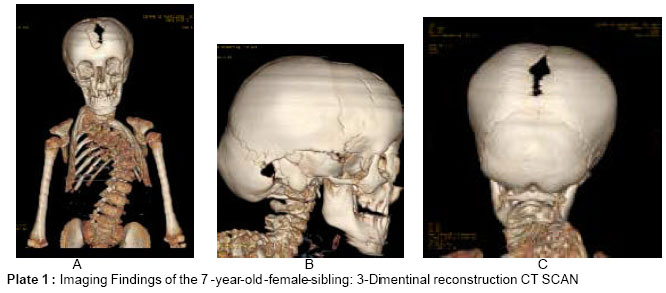
African Journal of Biomedical Research, Vol. 8, No. 2, 2005, pp.127-129 Case Report Massive spinal–ischial dysplasia and extensive spinal dyssygmentation in a Tunisian child with severe form of Cleido-cranial dysplasia Ali Al Kaissi[a]*, Farid B Chehida**, Hatem Safi*, Maher Ben, Ghachem*, Nabil Nassib*, Jalel Chakib*, and Hassan Gharbi** *Service
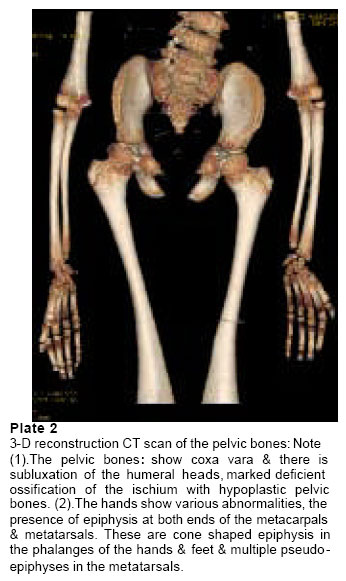
d’Orthopedie Infantile Hopital d’Enfants de Tunis Code Number: md05023 Abstract Kyphoscoliosis is a complication of some bone dysplasias, including Cleido-cranial dysplasia (CCD). We report on massive spinal dysplasia secondary to severe spinal dyssygmentation associated with marked defective ossification of the ischium, detected in a Tunisian female child with a severe form of Cleido-cranial dysplasia. Literature review on Cleido cranial dysplasia and associated spinal abnormalities showed no previous similar reports as encountered in our patient. Key words: Cleido cranial, dysplasia, spinal dyssygmentation, defective ossification of the ischium, kyphoscoliosis. INTRODUCTION Cleido-cranial dysplasia is a chondrodystrophic dysplasia characterised by defective skull ossification, clavicular hypoplasia or aplasia, delayed ossification of the pelvis, pseudoepiphyses and kyphoscoliosis, it is an autosomal dominant condition, however, the condition is classically characterised by deficient ossification of bones formed in membrane, though bones are formed by endochondral ossification can be affected. We report on a 7-year-old- female child manifesting the full blown and severe form of cleidocranial dysplasia, particularly her vicious kyphoscoliosis, the totally absent clavicles, and the remarkable defective ossification of the ischial rami, moreover, these malformation complex were remarkably demonstrated by means of 3-dimensional CT scan, since the standard radiographic examinations were not contributory. The older female, who is normally looking sibling, came to our department complaining of bouts of pain in her pelvic bones, when radiographs were taken we observed important dysplasia of her ischial rami, and discrete adolescent kyphosis as well. While further exploring this family we observed that the normally looking 43-year-old-mother is with the same pelvic dysplastic process. The purpose of this result is to illustrate the result of similar pathogenetic mechanisms in the same family, however, with different intensities. Once the mechanism is discovered in one family member, a search for similar mechanisms in others can be rewarding. CASE REPORT AND DISCUSSION The skull shows severe degrees of imperfect ossification of the sutures: 1. The anterior & posterior fontanelle is large and remarkably opened. 2. The nasal bones are absent with marked deficient dentition, extends from Inferior nasal downward. 3. The clavicles show total absence, bilaterally. 4 Spinal column: marked kyphoscoliosis, because of massive hemivertebrae extending from the cervical to the lumbo-sacral. 5. Deficient ribsnumber (10 ribs bilaterally), and the presence of the cervical rib and markedly hypoplastic scapulae. In this case study we are focusing on the following points for proper understanding of the inheritance pattern of any given skeletal dysplasia disorder in ascertained families, through; Firstly the knowledge of developmental patterns shared by different genetic disorders cautions the diagnostician and encourages a two-step procedure: a) provisional recognition of a pattern and b) more careful analysis of the pattern to reach a final, specific diagnosis. And secondly families of bone dysplasias may be the result of similar pathogenetic mechanisms, however, once the mechanism is discovered in one member of the family, a search for similar mechanisms in others may be rewarding. Hypoplasia of the ischia is an extremely rare congenital malformation, which has been reported as an isolated anomaly in very few cases. It has been known as a syndromic association complex in some congenital malformation syndromes, such as ischio-vertebral dysplasia, ischio-patellar dysplasia, and severe postaxial acrofacial dysostosis. We encountered three family members, in whom defective ossification of the ischium was noted, the index case, which manifested the full-blown picture of Cleido-cranial dysplasia, and her older female sibling and the mother, The reported condition is congenital & familial, with autosomal dominant inheritance and wide variation in expressions and variable clinical presentations in other normally looking subjects. The vast majority of reported cases of Cleido-cranial dysostosis are focusing on the congenital aplasia or hypoplasia of the clavicles, in our reported family we observed that the proband is the only child in the family with the most severe form of the disorder, however, older female sibling and her mother are complaining from pelvic pains and hip joints,radiographs showed small capital femoral epiphyses and development of post-adulthood arthrosis, discrete adolescent kyphosis and deficient ossification of the ischium in these normally looking family subjects. Ischio-vertebral dysplasia was encountered in this family with different grades and intensities, in the youngest female sibling who manifested the full blown picture of Cleido-cranial dysplasia, whereas the older female sibling and her mother were partially affected, however, their clavicles were normal and mostly the ischial rami in the older female child and her thoracic spines were affected, however, with less than what has been seen in the index case. The genetic basis for this syndrome has been well reported, Brueton et al., (1992) reported three apparent cases with re-arrangements of 8q22. A mother and daughter had a balanced t(8;10)(q22.3;p12.3) and an isolated girl had a dup(8q13.3->q22.1). Few reports have looked at the associated orthopaedic abnormalities in normal phenotype family subjects, in connection with the pathogenetic mechanism, and this in itself can be misleading for the treating physician, our report emphasizes on the necessity of examining siblings and other family subjects, once the recognition of a bone dysplasia disorder has been established in the family. Identifying the precise etiology should be of prime importance in dealing with any chronic illnesses, the intervening family members are variably affected, and their clinical presentations should not be separated in the management from the actual causation of their illnesses, the hip dysplasia of the mother and her female sibling is part of the syndromic association in this family. CONCLUSION Computed tomography is the investigation of choice to optimally demonstrate the massive bony abnormalities in our patient, however, when we applied the plain radiographs as our first modality, we felt that important radiological features are still missing, therefore, reconstruction was obtained and the fine details of this severe bony malformation complex was properly understood. We believe that etiology is the target in any research potential, and every ailment and malformation has a causative, and the connection to be made between onset of any given malformation and other similar mechanisms in families with recognized pathogenetic features, furthermore, if the cause is unknown, how can relevance be determined. REFERENCES
|

{kind=link}
{kind=link}