 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Malaysian Journal of Medical Sciences, Vol. 11, No. 1, January 2004, pp. 9-23 REVIEW ARTICLE Cell Targeting in Anit-Cancer Gene Therapy Mohd Azmi Mohd Lila, John Shia Kwong Siew, Hayati Zakaria*, Suria Mohd Saad*, Lim Shen Ni* and Jafri Malin Abdullah** Institute of Bioscience, *Faculty of Veterinary Medicine, Universiti
Putra Malaysia, 43400 UPM Serdang, Malaysia. *Department of Neuro Sciences,
School of Medical Sciences, Universiti Sains Malaysia 16150 Kubang Kerian,
Kelantan, Malaysia
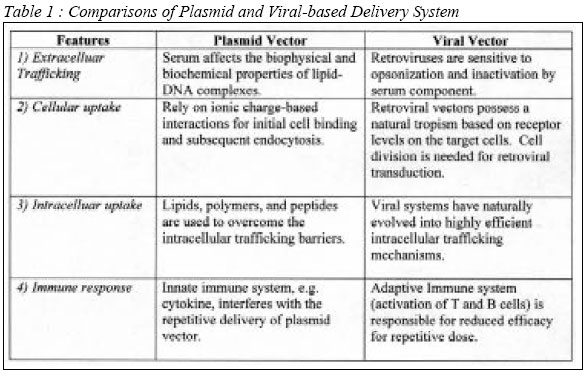
Code Number: mj04002 Gene therapy is a promising approach towards cancer treatment. The main aim of the therapy is to destroy cancer cells, usually by apoptotic mechanisms, and preserving others. However, its application has been hindered by many factors including poor cellular uptake, non-specific cell targeting and undesirable interferences with other genes or gene products. A variety of strategies exist to improve cellular uptake efficiency of gene-based therapies. This paper highlights advancements in gene therapy research and its application in relation to anti-cancer treatment. Key words : Gene therapy, anti-cancer, cell targeting, apoptosis Introduction Gene therapy is an advanced approach of medical intervention to correct inheritable genetic disorder through gene replacement. The goal for gene-based therapeutics is the effective delivery of genes to the diseased tissue or organ with subsequent expression of a therapeutic protein. Generally, the approach is intended to confer a therapeutic or prophylactic effect. However, it may serve as a way of marking cancer or any target cells for later identification. Its application is based primarily on modification of the genetic material of living cells. Therefore, the primary considerations for the design of gene delivery vehicle should include cellular internalization, intracellular trafficking, nuclear uptake, and the retention period of an expressed protein. Upon administration of suitable genetic materials to the subject or patient, the target cells are expected to be modified or altered in vivo. There arise several issues including the safety of the delivery system, the safety of the expressed gene product, and appropriate host-associated immune response. These problems can be minimized when an efficient targeting requirement is fulfilled. In principle, it requires an appropriate recombinant DNA material to carry and transfer the desired genetic material. This may involve extensive genetic manipulations and lengthy studies and explains why only a few gene therapy products have reached clinical trials. Gene therapy approaches vary from delivery of many copies of a gene, through gene modifications by using the properties of ribozymes, to injection of ex vivo modified cells. The ultimate goal of such approaches is to inactivate or to repair the mutated gene in the target cells or perhaps leading to elimination of such cells. However, the major challenge is not in the engineering of the target gene. Identifying an efficient vector and delivery method, regulating the transgene expression and maintaining the stability of gene expression are much more challenging. Viral vectors or non-viral delivery systems have been used in delivery of the therapeutic gene either ex vivo or in vivo. However, the nature of the target cells and the required levels and stability of gene expression seem to determine the choice of delivery vehicles and routes of administration. Other challenging areas include regulating the magnitude of immune responses to gene products as well as undesired or unavoidable immune responses to other proteins of the encoding vector. Gene Delivery The gene delivery systems, both viral- and plasmid-based materials, are used to produce a therapeutic protein in sufficient quantity at the appropriate site to elicit the desired biological responses. Some of the most common vehicles are viral vectors that consists of modified viral genomes carrying the gene of interest. Alternatively, naked plasmid-based vectors (1) or complexed with a variety of agents might be considered in the application. The most common complexing agents include cationic liposomes and condensing agents such as polyethylenemine (PEI) and poly L-lysine. Gene delivery vehicles must successfully traverse multiple barriers as they transit from the site of administration to their final destination, the nucleus of target cells (2, 3). Layered over these delivery barriers is the added complexity that each class of vehicle (adenoviral, retroviral, plasmid-based and etc.) has its own advantages that affect these potentially rate-limiting steps. To generate functional genetic-based therapeutics, there must be a clear understanding of the requirement pertinent to the route of administration and the capability (as well as limitations) of the chosen delivery vehicle. Barriers encountered in gene delivery depend on the route of administration. However, gene delivery can be classified temporally and spatially into four major steps: (1) extracellular trafficking, (2) uptake into target cells, (3) intracellular trafficking, and (4) the retention period of on expressed protein. All delivery vectors must have the ability to function at each of these steps. For instance, highly efficient DNA transport through the cytoplasm and into the nucleus is of little consequence if the delivery vehicle never reaches the target cells (Table 1). Therefore, gene-based delivery has focused on the mechanism of delivery and ways to overcome those delivery barriers (4, 5). Extracellular Trafficking Gene delivery vehicles will encounter a physiological milieu quite different from that presented under in vitro conditions. For instance, systemic intravenous (IV) administration is limited by several extracellular trafficking barriers. Delivery systems must gain stability within a complex array of serum proteins, and must be capable of avoiding clearance by the host immune system, namely by phagocytic cells and the reticuloendothelial system (RES). Immune clearance presents further considerations more specific for viral systems (6). For example, to abrogate the effect of neutralizing antibodies in vitro and in vivo against adenoviral vector, polyethylene glycol (PEG) and lipid conjugates are commonly used (7). In the case of plasmid vectors, it is believed that serum affects the biophysical and biochemical properties of lipid-DNA complexes. Serum alters both the size and charge, leading to an increase in complex disintegration, DNA release, and ultimately degradation (8). This implies that the delivery vehicles that arrive at the target site could differ substantially from that originally formulated and administered to the patient. Transfection efficiency may be affected by the composition of lipids in plasmid-based delivery systems (9, 10). The lipid composition is also an important factor in the recruitment of serum proteins (8, 10). There is also a direct relationship between the serum stability of lipid-DNA-based systems and their relative transfection efficiency. Therefore, understanding the subtle balance of aggregation, disassembly, and DNA degradation involved in gene delivery is crucial for development of gene therapy products. Although the interaction of plasmid delivery complex with serum components is unavoidable following intravenous administration, the ultimate effect in practical terms is less obvious. For instance, the deliverable complex may interact with serum complement proteins, but the interactions may not influence the transfection efficiency or systemic distribution of the complexes (11). Retroviruses delivery vehicles were also reported to be sensitive
to opsonization and inactivation by serum components following systemic delivery
(12). Furthermore, antigens from the packaging cell line used for the generation
of recombinant retrovirus may activate immune response and profoundly accelerate
viral clearance and reduce stability. Thus varlous, different packaging cell
lines are currently being investigated to minimize immune response activation
(13). The issue of in vivo stability becomes the limitation of the primary
usage of retroviruses in humans for ex vivo applications. However, direct in
vivo intratumoral administration of retroviral vectors has been accomplished
using the interferon- Targeting a specific disease requires knowledge of the appropriate tissue and cell types necessary to express the desired therapeutic protein. In addition, an understanding of the delivery system and route of delivery is also important to achieve the clinical goal. To date, biodistribution studies have established a foundation for IV delivery of lipid-DNA complexes (14, 15). Comparison of IV versus intratracheal (IT) plasmid-based delivery of the cystic fibrosis transductance regulator (CFTR) gene (16) demonstrated differential uptake and expression based on the delivery route. Following IT administration, DNA was found in the epithelial lining of the bronchioles (i.e. clara cells), whereas, following IV administration of a cationic lipid-based delivery system, DNA was delivered to the distal lung in the alveolar region, including alveolar type II epithelial cells (16). The lack of in vitro and in vivo correlation in gene expression profiles highlights another confounding issue of gene based delivery vehicles. For instance, blood components as the extracellular barriers for IV delivery are quite different from mucus barriers, which is relevant for IT or aerosol lung delivery. Obviously, the latter is the preffered delivery route for the CFTR gene (17), and the CF sputum has its own unique delivery barriers (18, 19). Due to the ease of administration through the nasal route, surface active agents or Dnase (19) has been applied to the airway passages to overcome CF sputum barrier for enhanced transfection by adenoviral and lipidic delivery systems. The coadministration of these agents will provide a more hospitable environment for the delivery system. Several other delivery strategies have been employed to avoid the potentially deleterious extracellular barriers of IV and IT delivery through direct administration of the gene delivery vehicle to the tissue of interest such as intratumoral injection (20), intramuscular injection (21-23) and particle bombardment via gene gun (24, 25). Electroporation, on the other hand, can enhance the transfection efficiency for the IM route (26). Cellular Uptake The importance of receptor levels was highlighted by analyses demonstrating the variability seen in gene expression levels (27) and distribution of adenovirus receptors (28, 29). As a consequence, decisions for adenoviral gene delivery strategies should be based on the assessment of receptor levels in target tissues, since variability in receptor availability could profoundly affect the outcome of adenovirus-based gene delivery. The major challenge now is how to compensate the receptor deficiency in the target tissue. Modifications of adenovirus fiber proteins will provide alternative cell-binding epitopes to retarget viral infectivity, which should help to alleviate limitations of viral delivery system based solely on receptor availability (30). In addition, this retargeting strategy provides an opportunity to generate the desired specificity for target cells. Without specificity, all cells possesing the coxsackievirus and adenovirus receptor (CAR) will be transduced by adenovirus. This may bring about the danger of potential toxicities due to the increased dosing needed to overcome this lack of specific targeting as well as potentially dangerous side effects due to the expression of the therapeutic gene in an inappropriate cell population. Specific targeting is especially desired for the delivery of therapeutic genes into tumor cells (31). The goal of retargeting is mainly to eliminate natural viral tropism while providing novel interaction. Several strategies, typically through antibodies (or fragments thereof), have shown promise in proof-of-principle studies for adenovirus (ad) as well as adeno-associated virus (AAV) (32, 33). These strategies have also enhanced transfection efficiency in cases where CAR is a limiting factor, and thus reduce toxicity associated with the increased dose of adenovirus required to achieve a therapeutic effect (34). Retroviral vectors, for example the prototype murine leukemia virus (MuLV), possess a natural tropism based on receptor levels on the target cells. Cells competent for retroviral transduction present the natural MuLV receptors and are actively dividing. Although cell division is a prerequisite for retroviral transduction (35), rapid proliferation alone is not sufficient for transduction efficiency. This implies that transduction efficiency may also be limited at the receptor level (36). To further support this hypothesis, as with adenovirus, increasing the pit2 receptors level from 18 000 to 150 000 per cell increased the MuLV transduction efficiency from 10 to 50% in rat 208F embryo fibroblast (37). Thus efficient cell transduction needs active cell division and an adequate number of receptors. In addition to the standard reengineering of genetic and biochemical functionalities of retrovirus, novel approaches have focused more on the physicochemical forces involved in retroviral-cell interaction (38). Physicochemical forces determine the binding of the retroviral vector to the target cell and the kinetic interplay between cell cycle and retroviral life cycle. This event will also determine the intracellular fate of the virus and ultimately constrain the efficiency of the gene transfer process (38). Plasmid-based systems rely on ionic charge-based interactions for initial cell binding and subsequent endocytosis (39), and the direct membrane fusion is the alternate route for cationic lipid-DNA-based systems. However, the contribution of these two pathways to nuclear delivery still remains unclear. The variation in serum interactions and transfection efficiency among existing lipid formulations makes it difficult to generalize regarding uptake mechanisms (9, 10). Furthermore, polymer-based systems show considerable differences compared to lipid systems. Targeting strategies for plasmid-based systems attempts to increase efficiency and specificity of delivery (40, 41). However, the major drawback of adding targeting elements or other protein components to plasmid-based systems to mimic the positive qualities of viral delivery systems, will be the increase of immunogenicity. Intracellular Uptake Viral systems have naturally evolved highly efficient intracellular trafficking mechanisms. And for this reason, viral systems have been used for gene delivery with minimal attempts to modify their inherent functionality. And again, adenovirus will be used as a typical example. Understanding the multiple functions afforded by the adenovirus coat protein in the intracellular phase of its infection cycle, has provided a foundation to engineer plasmid-based delivery systems. Construction of plasmid-based systems has focused on abrogating intracellular trafficking barriers such as endosomal entrapment and nuclear uptake (5, 42). Various lipids, polymers, and peptides have been employed in plasmid-based systems to overcome intracellular trafficking barriers (5, 39). Endosomal release and nuclear uptake are the primary foci of research for better transfection efficiency (i.e. expression) following entry of plasmid into the cell. Decomplexation and cytoplasmic transport are believed to be the secondary barriers. Increased efficiency of intracellular trafficking will result in an improved therapeutic index. This will allow reduced dosing of the plasmid delivery system to obtain the desired therapeutic effect, and consequently, this should also minimize potential toxicities related to high dosing (43). In order to overcome the deleterious effects of endosomal entrapment (5, 43, 44), several endosomolytic agents have been incorporated into plasmid-based systems (42, 45). Analogous to the retroviral systems (35), it requires cell proliferation for successful transfection (46). The hypothesis is that nuclear membrane breakdown during mitosis is required for uptake of plasmid DNA into the nucleus. In contrast, adenoviral vectors are able to transduce nondividing cells (47), suggesting that the viral genome had evolved a means to pass through the nuclear membrane. To date, a number of strategies are underway to overcome the nuclear membrane barrier for plasmid-based systems. For instance, incorporation of peptide nuclear localization signals (NLSs) into plasmid delivery systems helps transport plasmid into the nucleus of so-called quiescent cells (5, 48, 49). Alternatively, a nuclear transport cis-acting DNA element from SV40 may prove useful in plasmid-based systems (50, 51). T7 polymerase system utilizes cytoplasmic expression by circumventing the needs for nuclear transport. Increased expression of a marker gene in mouse brain tumors and direct intratumoral injection of the T7/tk gene (52) have demonstrated that the use of xenogenic protein (e.g. phage T7 polymerase) may raise potential immunogenicity issues. The cytoplasm of the cell presents transport and stability barriers to gene delivery regardless of the location of gene expression (i.e. nuclear or cytoplasmic). Obviously, the potential negative impact of DNA instability within the cytoplasm affects the efficiency of plasmid-based systems (53). Therefore, gene delivery agents must be capable of protecting the stability and integrity of the nucleic acid cargo during all stages of delivery. Apart from DNA loss due to nucleases and other degradative enzymes, the cytoplasm limits free diffusion of plasmid-sized molecules via its viscous environment (5, 54). Fortunately, adenovirus overcomes this environment by utilizing specific endogenous cellular molecular motors to facilitate transport through the cytoplasm (55). Persistent Gene Expression A good understanding in intracellular trafficking of DNA from cellular uptake through delivery into the nucleus should improve the efficiencies of gene transfer. Increased efficiencies of delivery and expression will ultimately affect dosing regimes, therapeutic indices, and safety profiles. However, the duration of gene expression and the impact of immunological responses directed against the delivery vehicle/or gene product are also important considerations for gene-based therapeutics. The desired level of persistence of therapeutic gene expression is variable for each specific therapeutic application. For instance, vaccination if given for long-term expression may have the potential danger of inducing immune tolerance. Similarly, expression of fast-acting cytokines with known systemic toxicities should be preferentially expressed in a controlled short-term manner. On the other hand, there is a need for sustained or repeated immunization in the treatment of established tumor. Persistent therapeutic protein gene expression can be achieved in one of two general ways: (1) a single dose of a persistently expressing vector or (2) repeat doses of less persistently expressing vector. Ad, AAV, retroviral, and plasmid-based gene delivery systems are the principal vectors being evaluated in a variety of animal and clinical models. Parameters for evaluation include their ability to express therapeutic proteins following administration by several routes, their persistence of expression, mechanisms limiting that persistence, and the ability to administer repeat doses. Innate immune responses take place immediately after injection, in eliminating the vast majority of recombinant Ad virions (6). However, in the case of first generation replication-deficient adenoviral vectors (E1-deleted or E1/E3-deleted), the onset of antigen-driven immunity directed against the remaining viral open reading frame (ORFs) is chiefly responsible for limiting their duration of expression (56-59). Loss of expression generally occurs by one month post-transduction. However, co-delivery of pharmacologic and vector-encoded immunosuppressive agents may prolonged its expression (60, 61). Alternatively, infection can be done in immunodeficient strains of mice (59, 61-63). These approaches have been important in determining the mechanisms limiting persistence and the ability to deliver vectors repetitively. The primary consideration now is the potential safety issues regarding the use of immunosuppressive agents in human gene-based therapeutics. Attempts to reduce vector immunogenicity can generate more persistent expression. For instance, the so called `gutless' Ad vector (41), has all its viral ORFs deleted, leaving only the transgene, the viral terminal repeats required for replication in packaging cells, a packaging sequence, and `stuffer DNA' to increase the genome length for packaging into viral capsids. Recombinant virus is produced by co-cultivation of a plasmid of the recombinant genome, together with a helper virus expressing all required trans-acting replication and packaging factors. These vectors demonstrate improved performance with respect to immunogenicity, safety, and duration of expression (52, 64). Gutless' AAV vectors bear only the transgene flanked by the viral inverted terminal repeats necessary for replication and packaging (65-67). Plasmids containing the recombinant viral genome or the replication and packaging proteins are transfected into tissue culture cells. These cells are then infected with adenovirus or transfected with the appropriate Ad gene expression plasmids to provide the helper functions necessary to initiate and sustain the replication and packaging of AAV virions. The duration of expression of AAV vectors introduced into the muscle of non-immunosuppressed animals can be in excess of several months (68). Similar to the `gutless' Ad and AAV recombinant vectors, plasmid-based vehicles encode only the desired therapeutic gene. In addition, most of the plasmid vectors lack protein components, which ameliorate the threat of antigen-specific immune responses that potentially shorten the duration of expression. In general, plasmid expression peaks at 8-12 hr following IV administration, and declines to undetectable levels after 2 to 3 days (8). The majority of plasmid-based systems exploit the human cytomegalovirus immediate-early (hCMV-IE) promoter-enhancer. This hCMV-IE promoter-enhancer has relatively lack of cell type specificity and high activity (69, 70). This sequence is used to express transgenes in recombinant adenoviral vectors. The expression will last for approximately one month only, post-IV administration, due to the interference of cell-mediated immune responses. The discrepancy between hCMV-IE promoter-enhancer persistence in plasmid-based and adenoviral vectors depicts different cellular responses to these vehicles. However, the promoter has been proven to be capable of persistent expression in the heterologous constructs. It is believed that some parts of the viral genome structure, not shared by most plasmids, may facilitate persistent gene expression. The IM route of plasmid-based gene delivery is notably different from that of IV route in its degree of persistentence. Administration of uncomplexed, "naked", or complexed DNAs by the IM route can result in prolonged expression greater than one year (71). The IM route provides a simple means of systemic expression, but may not be suitable for all applications. For instance, IM administration of the vascular endothelial growth factor (VEGF) gene resulted in substantial gene-dependent injury at the site of injection (72). Therefore, comparisons of IM route-dependent toxicity or pathology at the site of introduction are crucial, for each therapeutic protein-encoding vector. To enhance persistent gene expression, cis- and trans-acting viral DNA elements can be incorporated into plasmid vectors. For instance, sequences derived from viral replication origins and trans-acting origin binding factor ORFs derived from the Epstein-Barr Virus (EBV) (72, 73) and bovine papilloma virus (BPV) (74) genomes have been used. These plasmid vectors have part of the replication origin deleted, and thus prevented from DNA replication. The relative persistence of these vectors involves the interaction of the origin binding protein with both the plasmid-born replication origin sequence and the host cell chromatin, tethering the plasmid and preventing its loss during cell division (75). Unfortunately, a potential safety issue arises from the epidemiological association of EBV with several human cancers (76), and the demonstration that B-cell directed expression of the EBV origin binding protein, EBNA-1, in transgenic mice results in the development of lymphomas (77). The mutant EBNA-1 species is desirable, which retain the function of nuclear persistence while lacking oncogenic potential. This may be done through structure-function analyses if these two activities are indeed separable. The use of replicating vectors may help persistent gene expression. However, their use is associated with uncontrolled in vivo viral replication and is thus usually avoided. A modified replicating SV40-based plasmid system is under development (78). To minimize the risk of tumorigenesis, a specific set of mutations can be devised to reduce the oncogenicity of the large T antigen, without blocking its ability to bind to the origin. Alternatively, direct intratumoral injection can sufficiently limit undesirable distribution. The IM route is relatively useful for persistant in vivo expression applied to both plasmid-based and viral gene delivery systems. Additionally, the use of viral elements in plasmid-based vector gives long-term expression by other routes. However, in some gene therapeutics, prolonged expression is undesired due to potential adverse responses. Vector transcription could be regulated by administration of a short-lived nontoxic drug (79). In other words, inducers can be used to modulate the expression level of therapeutic. For example, regulation of transcription through the use of the insect hormone ecdysone (79), tetracycline (80), immunosuppressive rapamycin (2,81, 82), and the antiprogestins (83). Although promising, current outstanding issues include high basal levels of transcription in the absence of inducer, limited in vivo efficacy, and the possibility that these repressor proteins may be immunogenic, precluding repeat dose. Repeat Administration and Immune Activity Repetitive delivery of vectors may result in prolonged therapeutic expression of proteins in vivo. However, the effectiveness of subsequent dose is reduced under the influence of the innate and adaptive immune responses. Adenoviral vector is able to express efficiently on repeated doses only in nude mice or immunocompetent mice tolerized or transiently immunosuppressed by pharmacologic or vector-encoded agents (58, 59, 85-88). Unfortunately, even the `gutless' Ad vectors and AAV vectors have shown poor results in repeat delivery in non-immunosuppressed mice. These findings suggest that the capsid proteins associated with the incoming viral genomes are sufficient to generate a neutralizing immune response (89). `Serotype switching' is another mean to avoid adaptive immune responses. This involves the alternate use of recombinant viruses that are genetically divergent to avoid cross-neutralizing immune responses upon repeat delivery. Such an approach was tried out for both Ad (90, 91) and AAV (92, 93). Nevertheless, its application is still questionable due to the limited number of Ad and AAV serotypes available in cloned form. Plasmid-based vectors differs from viral systems in that
the innate immune system (i.e. cytokine), rather than the adaptive immune system
(T and B cell activation) appears to interfere with repetitive delivery. The
refractory period for productive IV delivery between the first and second dose
of plasmid vector ranges from 9-11 days (94). However, this refractory period
rely on the amount of the first treatment dose. The typical dose for plasmid
delivery is 50ug. However, when the interval between IV doses was set as 3
days, the initial dose should to be reduced to 0.5-5ug of liposome-complexed
DNA per mouse to allow subsequent gene expression. Delivery of liposome-complex
DNA via intraperitoneal route also showed a refractory period. Induction of
the refractory period is independent of expression protein expression or mRNA
by the first vector. Data showed that promoterless plasmid effectively induces
a refractory period of length similar to that of an expressing plasmid. Administration
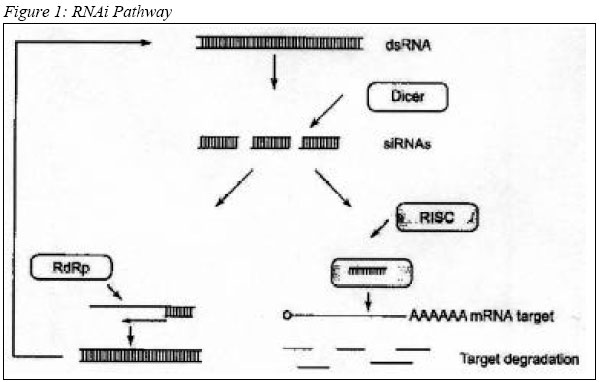
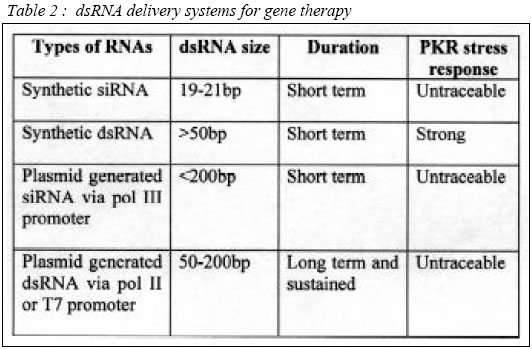
of liposome-complexed plasmid by the IV route elicits rapid IFN- Advances in Genetic-based Therapeutics RNA interference RNA interference (RNAi) methods are the most recent nucleic acid technology used for therapeutic purposes. The dsRNA activates a normal cellular process leading to a highly specific RNA degradation, and a cell-to-cell spreading of this gene silencing effect in several RNAi models (97). There are essentially three potential sites for therapeutic intervention: transcriptional, post-transcriptional, and post-translational. The RNAi pathway (Figure 1) begins with the cleavage of a dsRNA into 21-25bp small interfering RNAs (siRNA), by an RNaseIII-like enzyme called Dicer (98, 99). This 21-25bp siRNA species is then incorporated into multi-subunit RNA-induced silencing complex (RISC), which targets the unique cellular RNA transcript for enzymatic degradation. RNA hydrolysis occurs within the region of homology directed by the original siRNA (100), thereby selectively inhibiting target gene expression. The siRNA can also be used as primers for the generation of new dsRNA by RNA-dependent RNA polymerase (RdRp) (101). The systemic response in mice models RNAi-mediated gene silencing experiments were carried out in mice, by initiating a single tail vein injection of chemically synthesized siRNA probes. The results were variable ranging from 30-90% suppression of a stably integrated green fluorescent protein (GFP) transgene (102). The gene silencing encompassed most tissues analyzed, but unfortunately short lived, with a half-life of only 2-3 days. A single intramuscular injection of plasmid-based siRNA vectors co-injected with IL-12 in adult mouse have shown long-term in vivo gene silencing, exhibiting a duration of at least 120 days (103). dsRNA Delivery Strategies In general, dsRNA can be delivered in 3 ways: Chemically synthesized siRNA, siRNA expressing plasmid, and vector-based `large' dsRNA delivery. siRNAs used to generate the RNAi-mediated silencing can be synthesized chemically to closely mimic those found in vivo following the digestion of dsRNA by Dicer. These smaller siRNAs did not induce the dsRNA-dependent protein kinase (PKR) suppressive effects in contrast to their longer dsRNA relatives (100). siRNAs elicit transient RNAi responses in, Drosophila embyos (100), C. elegans (104, 105), and several mammalian cell lines (100, 106, 107). Unfortunately, the usefulness of siRNAs is limited by a relatively short and transient period of activity, particularly in human cells, and often a strong preference for certain sequences of the mRNA target for optimal activity (107). siRNA expressing plasmid vector is one of the current solutions to overcome the transient activity of exogenously added siRNAs. These custom-made plasmid vectors are incorporated with RNA polymerase III (pol III) promoters, such as U6 or H1, to allow for the intracellular expression of siRNAs (108-110). Transcripts derived from these promoters end in a run of 4-5 thymidines, which permits the specific determination of a transcript length, producing the effective siRNAs incorporated into the RISC complex. Intracellular expression of siRNAs from pol III promoters establishes an effective RNAi in mammalian cells. Levels of RNAi following intracellular expression in this manner appear to outperform the earlier exogenous administration of synthetic siRNAs. It permits a longer period of expression, particularly in cells where the expression unit becomes integrated into the host genome (108-112). Pol III system is notably an effective means to generate RNAi. However, its relatively small size limit of transcripts (113) could ultimately put an upper limit on the number of different siRNAs that can be generated from a single transcript. Different siRNA and siRNA-expressing plasmids have shown varying degree of RNAi from an identical target mRNA (107, 114). To date, vector-based `large' dsRNA delivery using Pol II or T7 promoter, is the most promising delivery method compared to the two methods discussed earlier on. RNA pol II-generated intracellular expression of a relatively large 500 nt hairpin-structured sRNA in mouse embyonic cell line induces RNAi and stable gene silencing in those cells (115). Longer dsRNA molecules (>50bp) have the advantage of presenting multiple Dicer-derived siRNAs to the cell. In other words, a longer dsRNA would permit targeting of more than one message with a single construct and could also potentially alleviate the development of resistance to potential RNAi therapies resulting from point mutations. Additionally, this allows the cell to employ the endogenous dsRNA silencing pathway to choose the most effective silencing siRNA(s). Plasmid-mediated expression of relatively long intracellular dsRNA in non-embryonic mammalian cells is able to induce efficient RNAi-mediated gene silencing for up to several weeks without eliciting generalized PKR-mediated suppressive response (116). Direct long dsRNA transfections were shown to elicit an anti-GFP RNAi response in zebra embryos. Furthermore, direct transfection have not been shown no stimulate the generalized suppressive PKR response seen in other cell lines and adult animals (27, 115). VP3 Protein of Chicken Anaemia Virus for Anticancer Gene Therapy VP3 is a small protein of 121 amino-acids with an estimated size of 16 kDa (116), derived from Chicken Anaemia Virus. VP3 sequence is unique in that it does not resemble any other sequenced animal or viral protein. VP3 is believed to serve as transcription regulator and/or DNA binding protein based on its proline rich content (117). Furthermore, the basic region, which is rich in proline, resembles nuclear localization signal or DNA binding domains (118) VP3 induces apoptosis in various human transformed and/or tumorigenic cell lines. Including, cell lines derived from hepatomas, lymphomas, leukemias, melanomas, breast and lung tumors, neuroblastomas, cholangio-, colon- and squamous cell carcinoma (119). The rate of VP3-induced apoptosis is variable in one tumor cell line to another, but it always reaches 90-100% apoptosis of the VP3-positive cells 6 days after transfection. VP3 protein is dispersed soon after synthesis throughout the nucleus and apoptosis occurred. VP3 aggregates and the cellular DNA condenses and/or is fragmented (119, 120). VP3 fails to induce apoptosis in normal lymphoid, dermal, fibroblastic, epidermal, endothelial and smooth-muscle cells in vitro. In addition, no apoptosis occurred upon its expression in rodent embryo diploid fibroblasts and hepatocytes. Long-term expression of VP3 in normal human fibroblast suggests that VP3 has no toxic or transforming activity in these cells (121). In tumor cells, VP3 is located in the nucleus, whereas in normal cells it was found in the cytoplasm. Thus, nuclear location is important for its apoptotic activity (122-124). Therefore, it is essential for VP3 to co-localize with chromatin for apoptotic event. The basic regions of VP3 allow interaction with nucleic acids. The presence of VP3 in the chromatin structure, together with its high proline content, may cause alteration of the supercoil organization, resulting in apoptosis. Another possibility is that VP3 acts as a transcriptional and/or inhibitor of genes, which directly mediate apoptosis (119, 120). (Table 2) Conclusion Gene therapy is a promising technology for treatment of cancer when it is genetically related. However, lack of understanding on the interactions between the protein expressed by the gene and the target gene has hindered rapid advancement towards clinical application. In addition, the effectiveness of most anti-cancer gene therapies relies on apoptosis, which involves a poorly understood and complex intracellular pathway. Furthermore, identification of an appropriate vehicle to transport "therapeutic genes" directly to cancer cells still remains as a great challenge to researchers. Although, some anti-cancer gene therapy products have reached clinical trials most faced critical problems at Phase II trials. These obstacles, however, should be treated as a great challenge towards development of useful anti-cancer gene therapy products in the near future. Acknowledgements The study is conducted as a collaboration between UPM and USM, and funded by National Cancer Council (MAKNA). References 1) Roland, A.P., From genes to gene medicines: Recent advances in nonviral gene delivery. Crit. Rev. Ther. Drug Carrier Syst. 1998; 15: 143-198 2) Magari, S. R., Rivera, V. M., luliucci, J. D., Gilman, M., and Cerasoli, F., Pharmacologic control of a humanized gene therapy system implanted into nude mice. J. Clin. Invest. 1997; 100: 2865-2872. 3) Mahato, R.I., Takakura, Y., and Hashida, M., Development of targeted delivery systems for nuclei acid drugs. J. Drug Target. 1997; 4:337-357 4) Lee, .R.J., and Huang, L., Lipidic vector system s for gene transfer. Crit. Rev. Ther. Drug Carrier syst. 1997; 14: 173-206. 5) Meyer, K.E.B., Uyechi, L.S., and Szoka, E.c., Manipulating the intracellular trafficking of nucleic acids. In: Gene therapy For Diseases of the Lung (K.L.brigham, ed.), pp135-180. Dekker, New York 1997. 6) Worgall, S., Wolff, G., falck-Pedersen, E., and Crystal, R.G., Innate immune mechanisms cominate elimination of adeniviral vectors following in vivo administration. Hum. Gen. Ther. 1997; 8: 37-44. 7) Cichon, G., and Strauss, M., Transient immunosuppression with 15-deoxyspergualin prolongs reporter gene expression and reduces humoral immune response after adenoviral gene transfer. Gene Ther. 1998; 5:85-90. 8) Zhao, D.D, Watarai, S, S., Lee, J.T., Kouchi, S., Ohmori, H., and and Yusuda, T., Gene transfection by cationic liposomes: Comparison of the transfection efficiency of liposomes prepared prepared from various positively charged lipids. Acta Med Okayama 1997 ; 51: 149-154 9) Li, S., Tseng, W.C., Beer Stolz, D., Wu, S.-P., Watkins, S.C., and Huang. L., Dynamic changes in the characteristics of cationic lipidic vectors after exposure to mouse serum: Implications for intravenous lipofection. Gene Ther. 1999; 6: 585-594. 10) Li, S.,Rizzo, M.A., Bhattacharya, S., and Huang, L., Characterization of cationic lipid-protamine-DNA (LPD) complexes for intravenous gene delivery. Gene Ther. 1998; 5: 930-937 11) Barron, L.G., Meyer, K.B., and Szoka, F.C., Jr., Effects of complement depletion on the pharmacokinetics and gene delivery mediated by cationic lipid-DNA complexes. Hum. Gene. Ther. 1998; 2: 151-155 12) Rother, R.P., Fodor, W.L., Springhorn, J.P., Birks, C.W., Setter, E., Sandrin, M.S., Suquinto, Sp.P., and Rollins, S.A., A novel mechanism of retrovirus inactivation in human serum mediated by anti-alpha-galactosyl natural antibody. J. Exp. Med. 1995; 182: 1345-1355 13) Pensierro, M.N. Wysocki, C.A., Nader, K., and Kikuchi, G.E., Development of amphotropic murine retrovirus vectors resistant to inactivation by human serum. Hum. Gene Ther. 1996; 7: 1095-1101 14) Mahato, R.I., Anwer, K., Tagliaferri, F., Meaney, C., Leonard, P., Wad, M.S., Logan, M., Frech, M., and Rolland, A., Biodistribution and gene expression of lipid/ plasmid complexes after systemic administration. Hum Gene. Ther. 1998; 9: 2083-2099 15) Niven, R., Pearlman, R., Wedeking, T., Mackeigan, J., Noker, P., Simpson-Herren, L., and Smith, J.G., Biodistribution of radio-labeled lipid-DNA complexes and DNA in mice. J. Pharm. Sci. 1998; 87: 1292-1299. 16) Grisenbach, U., Chonn,A., Cassady, R., Hannam, V., Ackerley, C., Post, M., Tanswell, A.K., Olek, K., O'Brodvich, H., and Tsui, L.C. Comparison between intratracheal and intravenous administration of liposome-DNA complexes for cystic fibrosis lung gene therapy. Gene Ther. 1998; 5:181-188 17) McDonald, R.J., Liggitt, H.D., Roche, L., Nguyen, H.T., Pearlman, R., Raabe, O.G., Bussey, L.B., and Gorman, C.M., Aerosol delivery of lipid: DNA complexes to lungs of rhesus monkeys. Pharm. Res. 1998; 15: 671-679 18) Kitson, C., Angel, B., Judd, D., Rothery, S., Severs, N.J., Dewar, A., Huang, L., Wadsworth, S.C., Cheng, S.H., Geddess, D.M., and Alton, E.W.E.W., The extra- and intracellular barriers to lipid and adenovirus-mediated pulmonary gene transfer in native sheep airway epithelium. Gene Ther. 1999; 6: 534-546 19) Stern, M., Caplen, N.J., Browning, J.E., Grisenbach, U., Sorgi, E., Huang, L., Gruenert, D.C., Marriot, C., Crystal, R.G., Geddes, D.M., and Alton, E., The effect of mucolytic agents on gene transfer across a CF sputum barrier in vitro. Gene Ther. 1999; 5: 91-98 20) Noruma, T., Yasuda, K., Yamada, T., Okamoto, S., Mahato, R.I., Wataneba, Y., Takakura, Y., and Hashida, M., Gene expression and antitumor effects following direct interferon (IFN)-gamma gene transfer with naked plasmid DNA and DC-chol liposome complexes in mice. Gene Ther. 1999; 6: 121-129 21) Kessler, P.D., Podsakoff, G.M., Chen, X., McQuiston, S.A., Colosi, P.c. P.C. Matelis, LA., Kurtzman, G.J. and Byrne, B.J., Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc. Natl. Acad. Sci. U.S.A. 1996; 93: 14082-14087 22) Marshall, D.J., and Leide, J.M. Recent advances in skeletal-muscle-based gene therapy. Curr. Opin, Genet. Dev. 1998; 8: 360-365 23) Monahan, P.E., Samulski, R.J., Tazelaar, J., Xiao, X., Nichols, T.C., Bellinger, D.A., read, M.S., and walsh, C.E., Direct intramuscular injection with recombinant AAV vectors results in sustained expression in a dog model of hemophila. Gene Ther. 1998; 5: 40-49 24) Mahvi, D.M., Sheehy, M.J., and Yang, N.S., DNA cancer vaccines: A gene gun approach. Immunol. Cell Biol. 1997; 75: 456-460. 25) Rakhmilevich, A.l., Timmins, J.G., Janssen, K., Poohmann, E.L., Sheehy, M.J., and Yang, N.S. Gene gun-mediated IL-12 gene therapy induces antitumor effects in the absence of toxicity: A direct comparison with systemic IL-12 protein therapy. J. Immunother. 1999; 22: 135-144 26) Mir, L.M., Bureau, M.E., Gelh, J., Rangra, R., Rouy, D., Caullaud, J.M., Delaere, P., Branellec, D., Schwartz, B., and Scherman, D., High efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc. Natl. Acad. Sci, U.S.A. 1999; 96: 4262-4267 27) Li, Y., Pong, R.V., Bergelson, J.M., Hall, M.C., Sagolowsky, A.I., Tsaeng, C.P., Wang, Z., and Hsieh, J.T., Loss of adenoviral receptor expression in human bladder cancer cells: A potential impact on the efficacy of gene therapy. Cancer Res. 1999; 59: 325-330 28) Nalbantoglu, J., Pari, G., Karpati, G., and Holland, P.C., Expression of the primary coxsakie and adenovirus receptor is downregulated during skeletal muscle maturation and limits the efficacy of adenovirus-mediated gene delivery to muscle cells. Hum. Gene Ther. 1999; 10: 1009-1019 29) Hemmi S, Geertsen R, Mezzacasa A, Peter I, Dummer R. The presence of human coxsackievirus and adenovirus receptor is associated with efficient adenovirus-mediated transgene expression in human melanoma cell cultures Hum. Gene Ther 1998; 9: 2363-73. 30) Hidaka, C., Milano, E., Leopold, P.L., Bergelson, J.M., Hackett, N.R., Finberg, R.W., Wickham, T.J., Kovesdi, I., Roelvink, P., and Crystal, R.G., CAR-cependent and CAR-independent pathways of adenovirus vector-mediat4ed gene transfer and expression in human fibroblasts. J. Clin. Invest. 1999; 103: 579-587 31) Bilbao, G., Gomez-Navarro, J., and Curiel, D.T., Targeted adenoviral vectors for cancer gene therapy. Adv. Exp. Med. Biol. 1998; 451: 365-374 32) Bartlett, J.S., Kleinschmidt, J., Boucher, R.C., and Samulski, R.J., Targeted adeno-associated virus vector transduction of nonpermissive cells mediated by bispecific F(Ab)2, antibody. Nat Biotechnol. 1999; 17:181-186. 33) Rogers, B.E., Douglas, J.T., Ahlem, C., Buchsbaum, D.J., Frincke, J., and Curiel, D.T., Use of a novel cross-linking method to modify adenovirus tropism. Gene Ther. 1997; 4: 1387-1392. 34) Rancourt, C., Rogers, B.E.,Sosnowski, B.A., Wang, M., Pichem A., Pierce, G.F., Alvarez, R.D., Siegal, G.P., Douglas, J.T., and Curiel, D.T., Basic Fibroblast growth factor enhancement of adenovirus-mediated delivery of the herpes simplex virus thymidine kinase gene results in augmented therapeutic benefit in a murine model of ovarian cancer. Clin. Cancer. Res. 1998; 4: 2455-2461. 35) Miller, D.G., Adam, M.A., and Miler, A.D. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell. Biol. 1990; 10: 4230-4242. 36) Orlic D, Girard LJ, Jordan CT, Anderson SM, Cline AP, Bodine DM. The level of MRNA encoding the amphotropic retrovirus receptor in mouse and human hematopoietic stem cells is low and correlates with the efficiency of rotavirus transduction Proc Natl Acad Sci. 1996; 93: 11097-102. 37) Kurre, P, Kiem, H. R, Morris, J., Heyward, S., Battini, J. L., and Miller, A. D., Efficient transduction by an amphotropic retrovirus vector is dependent on high-level expression of the cell surface virus receptor. J. Virol. 1999; 73: 495-500. 38) Palsson, B., and Andreadis, S., The physico-chemical factors that govern retrovirus-mediated gene transfer. Exp. Hematol. 1997; 25: 94-102. 39) Hope, M. J., Mui, B., Ansell, S., and Ahkong, Q. F., Cationic lipids, phosphatidylethanolamine and the intracellular delivery of polymeric, nucleic acid-based drugs. Mol. Membr. Biol. 1998; 15: 1-14. 40) Cristiano, R. J., Targeted, non-viral gene delivery for cancer gene therapy. Front. Biosci. 1998; 3:Dl 161-D1170. 41) Morsy, M. A., and Caskey, C. T., Expanded-capacity adenoviral vectors—The helper-dependent vectors. Mol. Med Today 1999; 5: 18-24. 42) Wagner, E., Effects of membrane-active agents in gene delivery. J. Controlled Release 1998; 53:155 -158 43) Zabner, J., Fasbender, A. J., Moninger, E, Poellinger, K. A., and Welsh, M. J., Cellular and molecular barriers to gene transfer by a cationic lipid. J. Biol. Chem. 1995; 270:18997-19007. 44) Brisson, M., He, Y., Li, S., Yang, J. P., and Huang, L., A novel T7 RNA polymerase autogene for efficient cytoplasmic expression of target genes. Gene Ther. 1999; 6:263—270. 45) Baru, M., Nahum, O., Jaaro, H., Sha'anani, J., and Nur, I. Lysosome-disrupting peptide increases the efficiency of in-vivo gene transfer by liposome-encapsulated DNA. J. Drug Target. 1998; 6: 191-199. 46) Fasbender, A., Zabner, J., Zeiher, B. G., and Welsh, M. J., A low rate of cell proliferation and reduced DNA uptake limit cationic lipid-mediated gene transfer to primary cultures of ciliated human airway epithelia. Gene Ther. 1997; 4: 1173-1180. 47) Greber.U. R, and Kasamatsu, H., Nuclear targeting of SV40 and adenovirus. Trends Cell Biol. 1996; 6: 89-195. 48) Ludtke, J. J., Zhang, G., Sebestyen, M. G., and Wolff, J. A., A nuclear localization signal can enhance both the nuclear transport and expression of 1 kbDNA. J. Cell Sci. 1999; 112: 2033-2041 49) Zanta, M. A., Belguise-Valladier, P., and Behr, J. P., Gene delivery: A single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:91-96. 50) Dean, D. A. Import of plasmic DNA into the nucleus is sequence specific. Exp. Cell Res. 1997; 230: 293-302. 51) Graessmann, M., Menne, J., Liebler, M., Graeber, I., and Graessmann, A., Helper activity for gene expression, a novel function of the SV40 enhancer. Nucleic Acids Res. 1989; 17: 6603-6612. 52) Chen, X., Li, Y., Xiong, K., Aizicovici, S., Xie, Y, Zhu, Q., Sturtz, E, Shulok, J., Snodgrass, R., Wagner, T. E., and Plan-ka, D., Cancer gene therapy by direct tumor injections of a nonviral T7 vector encoding a thymidine kinase gene. Hum. Gene Ther. 1998; 9: 729-736. 53) Lechardeur, D., Sohn, K.-J., Haardt, M., Joshi, P. B., Monck, M., Graham, R. W., Beatty, B., Squire, J., O'Brodovich,H., j and Lukacs, G. L., Metabolic instability of plasmid DNA in the cytosol: A potential barrier to gene transfer. J. Gene Ther. 1999; 6:482-497. 54) Luby-Phelps, K., Physical properties of cytoplasm. Curr. Opin. Cell Biol. 1994; 6: 3-9. 55) Suomalainen, M., Nakano, M. Y., Keller, S., Boucke, K., Stidwill, R. P., and Greber, U. E., Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. CellBiol. 1999; 144: 657-672. 56) Tripathy, S. K., Black, H. B., Goldwasser, E., and Leiden,]. M., Immune responses to transgene-encodedproteins limit the stability of gene expression after injection of replication-defective adenovirus vectors. Nat. Med. 1996; 2:545-550. 57) Yang, Y., Nunes, F. A., Berencsi, K., Gonczol, E., Engelhardt, J. E, and Wilson, J. M., Inactivation of E2a in recombinant adenoviruses improves the prospect for gene therapy in cystic fibrosis. Nat. Genet. 1994; 7: 362-369. 58) Yang, Y., Jooss, K. U., Su, Q., Ertl, H. C., and Wilson, J. M., Immune responses to viral antigens versus trans-gene product in the elimination of recombinant adenovirus-infected hepatocytes in vivo. Gene Ther. 1996; 3: 137-144. 59) Yang, Y., Greenough, K., and Wilson, J. M., Transient immune blockade prevents formation of neutralizing antibody to recombinant adenovirus and allows repeated gene transfer to mouse liver. Gene Ther. 1996; 3:412-420. 60) Bouvet, M., Fang, B., Ekmekcioglu, S., Ji, L., Bucana, C. D., Hamada, K., Grimm, E. A., and Roth, J. A., Suppression of the immune response to an adenovirus vector and enhancement of intratumoral transgene expression by low-dose etoposide. Gene Ther. 1998; 5: 189-195. 61) Elshami, A. A., Kucharczuk, J. C, Sterman, D. H., Smythe, W. R., Hwang, H. C, Amin, K. M., Lir/ky, L. A., Albelda, S. M., and Kaiser, L. R., The role of immunosuppression in the efficacy of cancer gene therapy using adenovirus transfer of the herpes simplex thymidine kinase gene. Ann. Surg. 1995; 222:298-307, 307-310. 62) Barr, D., Tubb, J., Ferguson, D., Scaria, A., Lieber, A., Wilson, C., Perkins, J., and Kay, M. A., Strain related variations in adenovirally mediated transgene expression from mouse hepatocytes in vivo: Comparisons between im-munocompetent and immunodeficient inbred strains. Gene Ther. 1995; 2: 151-155. 63) Zsengeller ZK, Wert SE, Hull WM, Hu X, Yei S, Trapnell BC, Whitsett JA. Persistence ofreplication deficient adenovirus-mediated gene transfer in lung of immune-deficient (nu/nu) mice. Hum Gene Ther, 1995; 6:457-67 64) Morsy, M. A., Gu, M. C., Zhao, J. Z., Holder, D. J., Rogers, I. T, Pouch, W. J., Motzel, S. L., Klein, H. J., Gupta, S. K., Liang, X., Tota, M. R., Rosenblum, C. I., and Caskey, C. T., Leptin gene therapy and daily protein administration: A comparative study in the ob/ob mouse. Gene Ther. 1998; 5: 8 -18. 65) Carter, B. J. Adeno-associated virus and AAV vectors for gene delivery. In "Gene Therapy: Therapeutic Mechanisms and Strategies" (D. Lasic and N. Templeton-Smith, eds.). Dekker, New York (in press) (2000). 66) Hallek, M., and Wendtner, C. M., Recombinant adeno-associated virus (rAAV) vectors for somatic gene therapy: Recent advances and potential clinical applications. Cytokines Mol. Ther. 1996; 2:69-79. 67) Rabinowitz, J. E., and Samulski, J.,Adeno-associated virus expression systems for gene transfer. Curr. Of in. Biotechnol. 1998; 9: 470-475. 68) Snyder, R. O., Spratt, S. K., Lagarde, C., Bohl, D., Kaspar, B., Sloan, B., Cohen, L. K., and Danos, O., Efficient and stable adeno-associated virus-mediated transduction in the skeletal muscle of adult immunocompetent mice. Hum. Gene Ther. 199; 8:1891-1900. 69) Miller, N., and Whelan, J. Progress in transcriptionally targeted and regulatable vectors for genetic therapy. Hum.GeneTher. 1997 8: 803-8l5. 70) Walther, W., and Stein, U. Cell type specific and inducible promoters for vectors in gene therapy as an approach for cell targeting. J. Mol. Med. 1996; 74: 379-392. 71) Wolff, J. A., Ludtke, J. J., Acsadi, G., William, P., and Jani, A., Long-term persistence of plasmid DNA and foreign gene expression in mouse muscle. Hum. Mol. Genet. 1992; 1: 363-369. 72) Guanghuan., T, Korchmaiet, A. L., Liggitt, H. D., Yu, W.-H., Heath, T. D., and Debs, R. J., Non-replicating EBV-based plasmids produce long-term gene expression in vivo. Submitted for publication 1999. 72) Springer, M. L., Chen, A. S., Kraft, P. E., Bednarski, M., and Blau, H. M., VEGF gene delivery to muscle: Potential role for vasculogenesis in adults. Mol. Cell 1998; 2: 549-558. 73) Sclimenti, C. R., and Calos, M. P., Epstein-Barr virus vectors for gene expression and transfer. Curr. Opin. Biotechnol. 1998; 9: 476-479. 74) Piirsoo, M., Ustav, E., Mandel, T, Stenlund, A., and Ustav, M., Cis and trans requirements for stable episomal maintenance of the BPV-1 replicator. EMBOJ. 1996; 15: 1-11. 75) Calos, M. P., Stability without a centromere. Proc. Natl. Acad. Sci. U.S.A. 1998; 95: 4084-4085. 76) Rickinson, A. B.,and Kieff, E. Epstein-Barr virus. In "Fields Virology" (B. N. Fields, D. M. Knipe, and P. M. How-ley, eds.), pp. 2397-2446. Lippincott-Raven, Philadelphia 1996. 77) Wilson, J. B., Bell, J. L., and Levine, A. J., Expression of Epstein-Barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. EMBOJ. 1996; 15: 3117-3126. 78) Cooper, M. J., Lippa, M., Payne, J. M., Hatzivassiliou, G., Reifenberg, E., Fayazi, B., Perales, J. C., Morrison, L. J.,Tem pleton, D., Piekarz, R. L., and Tan, J., Safety-modified episomal vectors for human gene therapy. Proc. Ntd Acad. Sci. U.S.A. 1997; 94:6450-6455. 79) Clackson, T., Controlling mammalian gene expression with small molecules. Curr. Opin. Ghem. Biol. 1997; 1: 210-218 79) No, D., Yao, T. P., and Evans, R. M., Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc. Natl. Acad. Set. U.S.A. 1996; 93: 3346-3351. 80) Blau, H. M., and Rossi, E M., Tet B or not tet B: Advances in tetracydine-inducible gene expression. Proc. NaL Acad. Sd. U.S.A. 1999; 96: 797-799, 81) Rivera, V. M., Clackson, T, Natesan, S., Pollock, R., Amara, J. E, Keenan, T, Magari, S. R., Phillips, T, Courage, N. L., Cerasoli, E, Jr., Holt, D. A., and Gilman, M., A humanized system for pharmacologic control of gene expression. Nat. Med. 1996; 2: 1028-1032. 82) Ye, X., Rivera, V. M., Zoltick, P., Cerasoli, E, Jr., Schnell, M. A., Gao, G., Hughes, J. V, Gilman, M., and Wilson, J. M., Regulated delivery of therapeutic proteins after in vivo somatic cell gene transfer. Science 1999; 283: 88-91. 83) Burcin, M. M., Schiedner, G., Kochanek, S.,Tsai, S. Y, and O'Malley, B. W., Adenovirus-mediated regulable target gene expression in vivo. Proc. Natl. Acad. Sci. U.S.A. 1999; 96: 355-360 84) Benihoud, K., Saggio, I., Opolon, P., Salone, B., Amiot, E, Connault, E., Chianale, C., Dautry, E, Yeh, P., and Perricaudet, M., Efficient, repeated adenovirus-mediated gene transfer in mice lacking both tumor necrosis factor alpha and lymphotoxin alpha. J. Virol. 1998; 72: 9514-9525. 85) Dong, J. Y., Wang, D., Van Ginkel, F. W., Pascual, D. W, and Frizzell, R. A., Systematic analysis of repeated gene delivery into animal lungs with a recombinant adenovirus vector. Hum. Gene Ther. 1996; 7: 319-331 86) Kagami, H., Atkinson, J. C., Michalek, S. M., Handelman, B., Yu, S., Baum, B. J., and O'Connell, B., Repetitive adenovirus administration to the parotid gland: Role of immunological barriers and induction of oral tolerance. Hum. Gene. Ther. 1998; 9: 305-313. 87) Smith LM, Birrer MJ. Use of transcription factors as agents and targets for drug development oncology (Hunlight) 1996; 10: 1532-8. 88) Yei, S., Mittereder, N.,Tang, K., O'Sullivan, C., and Trapnell, B. C., Adenovirus-mediated gene transfer for cystic fibrosis: Quantitative evaluation of repeated in vivo vector administration to the lung. Gene Ther. 1994; 1: 192-200. 89) Kafri, T, Morgan, D., Krahl, T, Sarvetnick, N., Sherman, L., and Verma, I., Cellular immune response to adenoviral vector infected cells does not require de novo viral gene expression: Implications for gene therapy. Proc. Natl Acad. Sci. U.S.A. 1998; 95: 11377-11382. 90) Kass-Eisler, A., Leinwand, L., Gall, J., Bloom, B., and Falck-Pedersen, E., Circumventing the immune response to I adenovirus-mediated gene therapy. Gene Ther. 1996; 3: 154- 162. 91) Roy, S., Shirley, P. S., McClelland, A., and Kaleko, M., Circumvention of immunity to the adenovirus major coat protein hexon. J. Virol. 1998; 72: 6875-6879 92) Rutledge, E. A., Halbert, C. L., and Russell, D. W., Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J. Virol. 1998; 72: 309-319. 93) Xiao, W., Chirmule, N., Berta, S. C., McCullough, B., Gao, G., and Wilson, J M., Gene therapy vectors based on adeno-associated virus type I. J. Virol. 1999; 73: 3994-4003. 94) Song, Y. K., Liu, E, Chu, S., and Liu, D., Characterization of cationic liposome-mediated gene transfer in vivo intravenous administration. Hum. Gene Ther. 1997; 8: 1585-1594. 95) Dow, S. W, Fradkin, L. G., Liggitt, H. D., Willson, A. E, Heath, T. D., and Potter, T. A., Lipid-DNA complexes induce potent activation of innate immune responses and and tumor activity when administered intravenously. J. Immunol. 1999; 163: 1552-1561. 96) Whitmore, M., Li, S., and Huang, L., LPD lipopolyplex initiates a potent cytokine response and inhibits tumor growth. Gene Therap. 1999; 6: 1867-1875 97) Winston W.M.; Molodowitch C.; Hunter C.P. Systemic RNAi in C. elegans requires the putatice transmembrane protein SID-1. Science 2002; 295: 2456-2459 98) Zamore, P.D., Tuschl, T., Sharp, P.A., Bartel, D.P., RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nuvleotide intervals. Cell 2000; 101: 25-33 97) Krieg, A. M., An innate immune defense mechanism based on the recognition of CpG motifs in miciobial DNA. J. Lab. Clin. Med. 1996; 128: 128-133. 98) Pisetsky, D. S., Immune activation by bacterial DNA: A new genetic code. Immunity 1996; 5: 303-310. 99) Hutvagner, G. and Zamore,P.D., RNAi: Nature abhors a bouble-strand. Curr. Opin. Genet. Dev. 2002; 12: 225-232 100) Elbashir, S.M., Lendeckel, W., Tuschl, T., RNA interference is mediated by 21- and 22- nucleotide RNAs. Genes Dev. 2001; 15: 188-200. 101) Ahlquist, P., RNA-dependent RNA polymerases, viruses, and RNA silencing. Science 2000; 296: 1270-1273 102) Lewis, D.L., Specific inhibition of gene expression in post-natal mammals using small interfering RNAs. Keystone symposia: RNA interference, cosuppression and related Phenomena, 2002, February 21-26. Abstract no. 312. 103) Pachuk, C.J., dsRNA mediated post-transcriptional gene silencing and the interferon response in human cells and an adult mouse model. Keystone symposia: RNA interference, co-suppression and related Phenomena, 2002, February 21-26. Abstact: 217 104) Caplen N.J.; Parrish S.; Imani F.; Fire A.; Morgan R.A., Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate system. Proc. Natl. Acad. Sci. U.S.A. 2001; 98: 9742-9747 105) Parrish, S., and Fire., A distinct roles for RDE-1 and RDE-4 during RNA interference in Caenorhabditis elegans. 2001; RNA 4: 1397-1402. 106) Yu, J.Y., DeRuiter, S.L., Turner, D.L., RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2002; 99: 6047-6052 107) Holen, T., Amarzguioui, M., Wiiger, M.T., Babaie, E., Prydz, H., Positional effects of short interfering RNAs targeting the human coagulation trigger tissue factor. Nuclei Acids Res. 2002; 30: 1757-1766. 108) Brummelkamp, T.R., Bernards. R., Agami, R., A system for stable expression of short interfering RNAs in mammalian cells. Science 2002; 296: 550-553. 110) Myslinski E.; Amé J.-C.; Krol A.; Carbon P. An unusually compact external promoter for RNA polymerase III transcription of the human H1RNA gene. Nucleic Acids Res. 2001; 29: 2502-2509 112) Paddison, P.J; Caudy, A.A; Hannon, G.J. Stable expression of gene expression by RNAi in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2002; 99: 1443-1448. 113) Miyagishi, M., and tiara, K. U6 promoter-driven siRNAs with four uridine 3' overhangs efficiently suppress targeted gene expression in mammalian cells. Nat. Biotechnol. 2002; 19: 497-500 114) Lee N.S.; Dohjima T.; Bauer G.; Li H.; Li M.-J.; Ehsani A.; Salvaterra P.; Rossi J. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol. 2002; 19: 500-505. 115) Paddison, P.J; Caudy, A.A; Bernstein, E; Hannon, G.J; Conklin, D.S. Short hairpin RNAs induce sequence specific silencing in mammalian cells. Genes. Dev., 2002; 16: 948-958 116) Pachuk, C.J., Ciccarelli, R.B., Samuel, M., Bayer, M.E., TroutmanJr,R.D., Zurawski, D.V. Schauer, J.I., Higgins, T.J., Weiner, D.B., Sosnoski, D.M., ZurawskiJr., V.R. and C. Satishchandran. Characterisation of a new class of DNA delivery complexes fromed by the local anesthetic bupivican. Biochim. Biophys. Acta 2001; 1468: 20-30 116) Noteborn, M.H.M., Van der Eb, A.J., Koch, G. and Jeurissen, S.H.M. VP3 of the chicken anemia virus (CAV) causes apoptosis. In Vaccines ed. H.S. Ginsberg., F. Brown., R.M. Chanock, and R.A. Lerner, pp299-304. Cold Spring Harbor, New York. Cold Spring Harbor Laboratory Press 1993. 117) Noteborn, M.H.M., Verschueren, C.A.J., Zantema, A., Koch, G. and Van der Eb, A.J. Identification of the promoter region of chicken anemia virus (CAV) containing anovel enhancer element. Gene, 1994; 150: 313-318. 118) Noteborn, M.H.M., Arnheiter, H., Richter-Mann, L., Browning, H. and Weissmann, C. Transport of the murine mx protein into the nucleus is dependent on a basic carboxyterminal sequence. Journal oflnterferon Research 1987; 7: 657-669. 119) Noteborn, M.H.M., Zhang, Y.-H., and Van der Eb, A.J. Apoptin8 specifically causes apoptosis in tumor cells and after UV-treatment in untransformed cells from cancer-prone individuals: A review. Mutation Research 1998; 400: 447-455. 120) Noteborn, M.H.M., Danen-Van Oorschot, A.A.A.M., and Van der Eb, A.J. Chicken anemia virus: Induction of apoptosis by a single protein of a single-stranded DNA virus. Sem. Virol. 1998; 8: 497-504. 121) Danen-Van Oorschot. A.A., Fischer, D.F., Grimbergen, J.M., Klein, B., Zhuang. S., Falkenburg, J.H., Backendorf, C., Quax, P.H., Van der Eb, A.J., Noteborn, M.H. Apoptin induces apoptosis in human transformed and malignant cells but not in normal cells. Proc. Natl. Acad. Sci. ISA. 1997; 94: 5843-5847 122) Zhuang, S.M., Landegent, J.E., Verschueren, C.A.J., Falkenburg, J.H.F., Van Ormondt, R, Van der Eb, A.J. & Noteborn. M.H.M. Apoptm. a protein encoded by chicken anemia virus, induces cell death in various human hematologic malignant cells in vitro, leukemia 1995; 9: SI 18-S120 123) Zhuang, S.M., Shvarts, A., Jochemsen, A.G., Van Oorschot, A.A.A.M., Van der Eb, A.J. and Noteborn, M.H.M. Differential sensitivity-to Ad5 ElB-21kD and Bcl-2 proteins of apoptin-induced versus p53-induced apoptosis. Carcinogene.sis 1995; 16: 2939-2944 124) Zhuang, S.M., Shvarts, A., Van Ormondt, H., Jochemsen, A.G., Van der Eb, A.J. and Noteborn, M.H.M. Apoptm. a protein derived from chicken anemia virus, induces p53-independent apoptosis in human osteosarcoma cells. Cancer Res. 1995; 55: 486-489 Copyright 2004 - Malaysian Journal of Medical Sciences The following images related to this document are available:Photo images[mj04002t2.jpg] [mj04002t1.jpg] [mj04002f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}