 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
REVIEW ARTICLE THE GENETICS OF SCHIZOPHRENIA Mohd Razali Salleh Human Genome Research Group,
School of Medical Sciences, Universiti Sains Malaysia Health Campus,
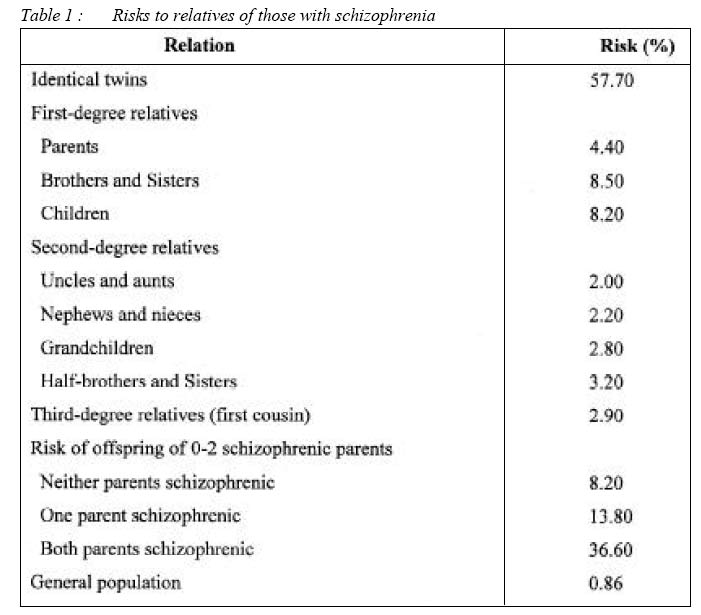
16150 Kubang Kerian, Kelantan, Malaysia Code Number: mj04014 ABSTRACT Schizophrenia is a complex biological disorder with multifactorial mode of transmission where non-genetic determinants are also play important role. It is now clear that it involves combined effect of many genes, each conferring a small increase in liability to the illness. Thus no causal disease genes or single gene of major effects, only susceptible genes are operating. Given this complexity, it comes as no surprise of the difficulty to find susceptible genes. However, schizophrenia genes have been found at last. Recent studies on molecular genetics of schizophrenia which focused on positional and functional candidate genes postulated to be associated with schizophrenia are beginning to produce findings of great interest. These include neuregulin (NRG-1, 8p12-21), dysbindin, (DTNBP1,6p22.3), G72 (13q34) / D-amino acid oxidase (DAAO,12q24), proline dehydrogenase (PRODH2, 22q11.21), catechol-O-methyltransferase (COMT, 22q11.21), regulator of G protein signaling (RGS-4), 5HT2A and dopamine D3 receptor (DRD3). Applications of microarrays methods were able to locate positional candidate genes related to dopaminergic, serotonergic and glutamatergic neurotransmission. New genome scan project, seen in the light of previous scans, provide support for schizophrenia candidate region on chromosome 1q, 2q, 5q, 6p, 8p, 10p, 13q,15q and 22q. Other reports described including the application of LD mapping and positional cloning technique, microarray technology and efforts to develop quantitative phenotype. More exciting finding is expected in near future with the completion of Hap Map project. Key words : schizophrenia, genetics, candidate gene, association study, linkage analysis Introduction Schizophrenia is the most common serious mental illness. It is characterized by disturbances in behaviour, thought, perception and lost of reality testing. The incidence of schizophrenia world-wide is between 15-20 per 100,000 per year; while the prevalence is about 0.5-1% with lifetime risk 0.9% (1). Majority of those affected will develop chronicity with frequent relapse and admission, while some of them will suffer for the whole life; about 10-15% of the schizophrenic patients will be dependent on others or need long-term institutional care. The illness causes high morbidity and great lost to the affected family and nation as a whole because majority of the patients started the illness in the productive age group, adolescent and young adults. It imposes a burden not only on the patient, but also carers, the health service and wider society; and certainly an expensive illness to treat The basic genetics of schizophrenia Schizophrenia aggregates in the families with no known familial subtypes. Twin and adoption studies (2) have shown that this familiarity is explained predominantly by genetic (vs. environment) factors, with estimates of genetic contribution ranging from 60 to 80%. However, these data do not follow a simple recessive or dominant pattern. If it were simple recessive, the frequency in children of two schizophrenic parents would be 100%, but is actually under 40%; if it were simple dominant, 50% of the offspring of one schizophrenic parent would be affected (actually 8-15%) and each person with schizophrenia would have one ill parent (actually in many cases both parents are well)(3). Moreover, the prevalence in offspring is too low to be consistent with the high (50% or more) monozygotic twin concordance rate. Thus, the genetic effect is not completely penetrant (i.e. 100% phenotype expression rate) indicating that many relatives of people with schizophrenia may carry silent genetic susceptibility. Detail of the risk of illness among relatives with schizophrenia is shown in table I. A further complication is the epidemiological evidence that, while a high population prevalence has been maintained, the reproductive rate of people with schizophrenia is low. As a consequence of this complex picture, the exact nature of genetic transmission has remained unclear. Currently, the most plausible hypothesis is that most cases of schizophrenia result from polygenic mechanism (a few or a larger number of interacting gene) probably interacting with a variety of non-genetic factors which may include in-utero viral infection, subtle birth trauma and drug abuse. Current methods for genetic analysis of complex disorder
Available methods generally rely on the analysis of DNA markers in subject to determine whether the distribution of marker alleles in some way predicts the presence of disease. Some studies consider DNA variations within or near ‘candidate gene’. Many groups are now examining ‘maps’of the DNA markers throughout the genome to identify chromosomal regions likely to contain loci that have a major influence on susceptibility. These studies rely on the phenomenon of ‘linkage’, as exemplified by the fact that when ill parent transmits a disease-causing allele to children, numerous alleles at nearby loci (such as DNA marker loci) are also transmitted because no recombination event has occurred in the region (4). Pedigree studies: LOD score methods
In pedigrees with multiple ill individuals, the LOD score method can be used
to determine whether the distribution of DNA markers alleles within each
pedigree predicts presence of disease. The LOD score (which is the logarithm
of the ratio of the likelihood of the observed marker genotype assuming
linkage and their likelihood assuming no linkage) is based on a mathematical
model of the mode of inheritance (how common is the disease allele, and
how likely is disease if someone has 0,1 or 2 disease alleles?). Our
uncertainty of the precise mode of inheritance in complex genetic disorder
such as schizophrenia introduce the possibility of mis-specifying the
model, resulting in inappropriate linkage data. Pedigree studies: allele-sharing (non-parametric) methods
The affected sib pair (ASP) method depends on the fact that when two siblings inherit alleles at a given locus, by chance alone they will share the same two allele 25% of the time, one of the same alleles 50% of the time, and share no alleles 25% of the time. If both siblings are afflicted with the same disease and there is linkage, ill siblings will share marker alleles more frequently. ASP tests analyzed the proportion of shared alleles and its statistical probability in a sample of affected sibling pairs. An alternative is the affected pedigree member (APM) method, which considers how often each pair of the affected relatives of any relationship share marker alleles versus the ‘by chance’expectation. However, AMP tests are less frequently used in schizophrenia linkages studies as compared with LOD score and ASP test. Linkage analysis
In its simplest form, linkage analysis aims to identify a chromosome segment that is inherited from the parents by all of the affected and none of the unaffected members in the family. These often involve the use of a series of polymorphic markers to screen the entire genome in groups of families or pairs of affected siblings. The part of a chromosome that is transmitted with the disease phenotype down each generation is the likely location of a disease related gene. Further efforts to characterize the gene can then focus on that region. Typically, linkage analysis uses families with a high incidence of disorder over two or more generations, or large cohorts of pairs of siblings, both affected by illness. Parametric linkage methods typically involve multigenerational pedigrees and require estimation of the mode of inheritance, gene frequencies, phenocopy rate, and penetrance of the disease genes. Nonparametric techniques such as the affected sibling pair approach have attracted more interest in schizophrenia genetic studies because they are model-free and more suitable for studies of complex diseases. Linkage analysis offers a more general approach for localizing predisposing genes. This strategy was recognized early in psychiatry. A hypothesis-free approach for mapping genes is required for linkage strategy. The first linkage studies in psychiatry, which was published in 1969 (5), focused on specific chromosome regions and used clinical genetic markers (monogenic disease with known localization). In subsequent years, clinical genetic markers were still used exclusively in this field (e.g. monogenic diseases such as red-green blindness or for the activity variability of certain enzymes). However, in the early 1980s, DNA markers (polymorphic sites with known position in the genome) were first applied to unravel the genetic basis of psychiatric disorders. Lately, informative marker systems spanning the whole genome; genome-wide scans are available (6). Association studies
Allelic association (or linkage disequilibrium) studies complement linkage and are another important method to locate genes. A well–known example of allelic association is the increased frequency of specific HLA antigens in several diseases. When a disease is transmitted from parent to offspring, the disease causing gene is inherited along with a segment of chromosome lying alongside. The pattern of DNA variants (polymorphism) within the segment will also be transmitted and all descendents who inherit the disease from a founding individual will inherit some of the surrounding haplotype. Markers very close to the gene will show allelic association with the disease in a population that includes the descendants of the disease founder. Since meiotic recombination at each generation steadily erodes the transmitted haplotype, only markers very close to a disease gene continue to show association over short genetic distances. The simplest design is to compare the frequency of alleles of a polymorphic marker in cases and control drawn from the same population. Association is particularly suited to the study of candidate genes considered on theoretical grounds to be implicated in disease process. The ideal situation for detecting association is when the polymorphism itself directly influences the function of a candidate gene. Association studies represent a powerful method to detect genes of small effect.
In case control association studies, the frequency of genetic variant
is compared in a group of affected individuals versus a group of individuals
unaffected with the phenotype of interest. This design is extremely
powerful and sensitive to difference in ethnicity between groups, which
may produce false positive (or negative) results (7). Family-based
association studies using designs such as the Transmission Disequilibrium
Test (TDT) compare the frequency of transmission of candidate alleles
from heterozygous parents with the frequency of non-transmission of
candidates alleles. Other variants of this approach include sibling-based
paradigms involving comparison of candidate allele frequencies between
affected individuals and their unaffected siblings. Family based approaches
are generally less powerful than case-control designs but offer advantage
of protecting against ethnic stratification (8). Linkage disequilibrium (LD) mapping and positional cloning
Positional cloning is the primary strategy available for finding susceptibility genes for disorders with unknown pathophysiology. There are four stages involved in the whole process: genome scan, fine-mapping, LD mapping and functional genomics. Firstly multiple affected families are screened with a DNA marker map of all chromosomes (genome scan) and followed by fine-mapping; in these candidate region, additional markers are genotyped 1-2 cM apart. The third stage is LD mapping to find the association of small sets of adjacent markers with disease, implicating one or two specific genes. Lastly functional genomics or physiological studies on animal model was initiated to establish the role of the gene and protein, effects of mutation on physiology and behaviour, and response to treatment for the disease. Quantitative traits analysis
Complex disorders can be conveniently considered as either qualitative or quantitative traits. Qualitative traits include all clinical diagnoses such as schizophrenia, affective disorder and major depression that enable individuals in a family or population to be categorized as affected and unaffected. The methods of linkage and association described before can be applied to these dichotomous variables. Quantitative traits include attributes such as height of person or schizophrenia-related vulnerability traits such as neuropsychological measures, neurological signs and clinical dimension. These are continuous variables considered to reflex the concerted action of many genes each having a small effect on the trait. A variety of methods have been developed to map these genes. Microarray technology
Recent advances in technology, including high-throughput methods such as microarrays, allow the screening of tens of thousands of genes (up to 30,000) in human in a relatively short period of time (9). Microarray strategies offer additional tool to identify genes or pathways for new and unique drug target, determine premorbid diagnosis, predict drug responsiveness for individual patients and eventually initiate gene therapy and prevention strategies. Molecular genetic studies, in combination with the extensive new body of sequence information for the human genome, are revolutionizing the way in which cellular processes are investigated. High-density microarrays allow the parallel and quantitative investigation of complex mixtures of RNA and DNA The central genetic dogma states that genomic DNA is first transcribed into mRNA, after which mRNA is translated into protein. Proteins are critical to a wide range of intra- and extracellular activities, including enzymatic, regulatory and structural function. Microarrays monitor the transcriptome, the collection of mRNA in a cell. Estimates suggest that 50% of human transcriptome is expressed in the brain (10). Changes in mRNA expression can, but not always, result in phenotypical and morphological differences. Alteration in patterns of expression of multiple genes can offer new data concerning regulatory mechanisms and chemical pathways. Novel genes and pathways that have never been link to the pathophysiology of psychiatric illness can emerge from microarray studies to provide new insight into the disease process and potential unique therapeutic drug targets. How the search began?
Several strategies are available in the search for the study goal. The search
for relevant DNA sequence variations can either focus on specific genes
with known physiologically relevant products (enzyme and other proteins)
or can precede in a hypothesis-free manner in which each gene is considered
in an a priori way as a putative candidate. The preferred search strategy
for the first approach involves association studies (family-based or
case-control samples) with candidate genes, which were selected on the
basis of the current pathophysiological knowledge; for the second approach,
linkages studies (particularly with a genome-wide approach) are required.
The latter approach was enormously successful in the study of monogenic
disorders. The systematic hypothesis-free genome-wide strategy required the following: systems of positional DNA markers placed densely on the whole genome (initially restrictive fragment polymorphism, then repeat length polymorphism and now single nucleotide polymorphisms markers); and sample of genetically informative families each with more than one affected case (e.g. extended pedigree with multiple cases or pair of affected siblings). Unfortunately, it has been shown that this strategy did not guarantee quick success for complex disorders (in psychiatry as well as in other areas of medicine) as contrast with monogenic diseases (6) Current issues and challenge.
The history of the search for genes contributing liability to schizophrenia is around a quarter of century old, but it is always dashed with nonreplication of the finding. This has been so despite consistent evidence from family, twin and adoption studies of an important genetic contribution; the heritability (or proportion of variance in liability explained by additive genetic effects) of schizophrenia is estimated to be approximately 80% (11). The reasons for the difficulty in finding genes include the complexity of the phenotype, heterogeneity and lack of biological marker. The mode of transmission is multifactorial where non-genetic determinants are also operating. As has been pointed earlier, schizophrenia does not conform to a classical Mendelian pattern of inheritance and it is now clear that most, perhaps all, cases involve the combined effects of many genes, each conferring a small increase in liability to the disorder; not due to single gene of major effects. As a consequence, a single gene does not seem to cause the disorder; thus no causal disease genes, only susceptibility genes are operating. Otherwise a consistently replicable linkage signal should have been detected. Advancement has also been hampered by the relatively small size of many studies. Not only are large sample needed to detect small effects, but even larger samples are needed to replicate positive findings One way of trying to overcome this problem is to combine the data from multiple studies. Several groups that have formed collaborative studies; and funding agencies such as the National Institute of Health (USA) are requesting that DNA samples be made available to all qualified investigators. Combining data from families selected in differing ways, from heterogenous ethnic population, using different markers for their genome scans and different methods of analyzing is problematic. Badner and Gershon (12) adapted a method originally proposed by R.A Fisher in which P–value from multiple studies may be combined after correcting each value from the size of the region containing the minimum P-value. An alternative approach used by Levinson et. al (13) divided the genome into segments or ‘bins’and used a ranking method to assess the degree of support across studies for linkage within each segment. The two methods yielded overlapping but somewhat different results. There are several weaknesses in the association studies. Even if an association between a variant of candidate gene and disease is found, it is difficult to interpret for the following reasons: First, association studies focus on one among a large number of candidate genes (or might even proceed genome-wide); this is usually a case of multiple testing, and appropriately adjusted levels of significant have to be applied. Criteria for handling this source of false-positive result remain to be agreed. Second, cases and control have to be comparable by population-genetic background. It is difficult to ensure that this criterion is met just by adopting careful sampling methods. Third, once an association is found, it is difficult to decide between two probabilities: (a) that the marker allele itself impacts on the risk for the disease, or (b) a genetic variant near to the marker allele is the real determinant and is in linkage disequilibrium with the disease allele. Only functional studies are suitable to resolve this ambiguity. In dealing with complex disorder such as schizophrenia, linkage analysis also
has inherent difficulties such as: (i) The magnitude of gene effect.
Given the results of genome scans in psychiatric disorders, the susceptibility
genes are likely to contribute to small or modest effects. So far,
linkage analysis has been enormously successful in detecting causal
or major gene effects, but not small effects; whereas association studies
are substantially more powerful in detecting minor or modest gene effects. (ii) Strength of the magnitude of linkage signals across populations. Some linkage
signals turned out to be replicable only in comparable genetic backgrounds,
and not in other populations; for instance in schizophrenia, some linkage
findings on 8p, 9q, 15q were replicable exclusively in African populations;
whereas that on 10p has so far been replicable only among Caucasian
populations (14). In view of serious inherent limitations of linkage analysis in complex diseases which was discussed; it has been questioned whether even the application of new and most informative marker systems will enable researchers to localize susceptible genes with modest effects by using the linkage strategy. Apparently the sceptical attitudes to the utility of linkage analysis in complex diseases are receiving more acceptance. Consequently, alternatives to linkage analyses are attracting growing attention. In particular, given the availability of densely placed marker systems as single nucleotide polymorphisms (SNPs) and of high-throughput techniques; genome-wide case-control association studies for a hypothesis-free search for susceptibility genes are better option. Theoretically, this linkage-equilibrium-based approach can be expected to reveal increased power, compared to linkage studies in detecting modest gene effects (odd ratio of up to 2) (6). Given this complexity, it comes as no surprise of the difficulty to find susceptible genes in schizophrenia. However, schizophrenia genes have been found at last. A potentially exciting phase of research is imminent. Linkage disequilibrium (LD) mapping studies are beginning to produce findings of great interest in some of these regions; and additional findings should be expected. Some of the recent findings will be discussed here. Recent Findings
When scrutinized new findings in molecular genetics of schizophrenia, particularly the progress since middle 2002, Elkin et al (16) found that several positional genes have received a good deal of attention. These include neuregulin (NRG-1, 8p21p12), dysbindin (DTNPB1,6p22.3), G72 (13q34)/ D-amino acid oxidase (DAAO, 12q24), proline dehydrogenase (PRODH2, 22q11.21), catechol-Omethyltransferase (COMT, 22q11.21) and regulator of G protein signaling (RGS-4,1q21-q22). The role of functional candidate genes, 5HT2A and dopamine D3 receptor (DRD3) have also been identified. Neuregulin
The findings so far regarding the role of NRG1 in schizophrenia are wholly consistent. All published studies to date support an association between NRG-1 and schizophrenia, but the functional significance of the risk haplotype is not yet known. There are, however several clues as to how NRG-1 contributes to illness. NRG-1 has a role in expression and activation of glutamate and other neurotransmitter receptors as well as role in neurodevelopment, affecting cellular differentiation and neuronal migration. Post-mortem brain studies have shown that the ErbB3 gene, which is of the family of neuregulin receptors, is down regulated in those with schizophrenia (17) Dysbindin
The evidence for the dysbindin gene having a role in the etiology of schizophrenia is also turning out to be generally consistent. The gene is located within a linkage region previously identified by the same group on chromosome 6p22.3 It codes for a protein which binds to dystrobrevin, part of the dystrophin receptor complex, involved in the pathogenesis of muscular dystrophy. The protein is also found in a small subset of axons (18). Some of these axons localize to anatomical regions implicated in schizophrenia and are postulated to be involved in synaptic formation and maintenance, signal transduction and receptor gene expression. This may be via N-methyl-D-aspartate (NMDA) receptor functioning G72 and D-amino acid oxidase.
A novel gene encoding for a protein called G72 was found to be associated with
schizophrenia by Chumankov et al in 2002 (19). It has been suggested
that these genes confer their increase risk for schizophrenia via glutaminergic
transmission. It is hypothesized that those who produced D72 exhibit
a lower NMDA-type receptor glutamate receptor activity. This would predispose
individual to develop schizophrenia via glutamate signaling hypofunction.
This may play a role in the neurodevelopment hypothesis of schizophrenia,
since NMDA receptor is critical for the development and modifiability
of neuronal contacts. The specific expression of G72 and DAAO may have
an important role in the prenatal and postnatal period. Interestingly,
G72 has also recently been implicated in bipolar disorder (20). It was
further found that an in-vitro interaction occurs between G72, DAAO and
another protein expressed in the human brain. A DAAO gene polymorphism
was itself shown to be associated with schizophrenia and interestingly,
combination of G72/DAAO genotypes was found to have an epistatic (or
interactive) effect on disease risk Protein dehydrogenase
PRODH is another positional candidate for schizophrenia. It is a mitochondrial enzyme involved in transferring redox potential across the mitochodrial membrane, first identified by Liu et al (21) in 2002. It was found on a 1.5 Mb region on chromosome 22q11, a region previously implicated in the etiology of schizophrenia both by linkage studies and by work on velo-cardia facial syndrome (VCFS) (22). In this disorder the genetic defect is a microdeletion of the 22q11 regions, and sufferers have a 20-30% chance of a schizophrenia-like syndrome (23). Subsequent studies by other researchers gave conflicting results. Clearly, more work is needed to determine the role of this gene in schizophrenia. Catechol-O-methyltransferase
Although numerous linkage and association studies have been carried out on this candidate gene, but until 2002 these failed to produce any conclusive results. Then a very large case-control study, including over 700 probands and 4000 controls, by Shifman et al (24) found a significant association between schizophrenia and COMT haplotype in Ashkenazi Jews. They suggested that the Val/Met polymorphism had only a moderate or no effect on schizophrenia risk and that more than one functional polymorphism within the COMT gene may be responsible. COMT codes for a gene product that inactivates catecholamines including dopamine. It does so by methylating m-hydroxy groups. Like PRODH, this gene is located in the 22q11 linkage/ VCFS microdeletion region, and is therefore a prime candidate gene to be involved in the risk of developing schizophrenia. Regulator of G protein signaling
Attention focused on RGS-4 following a microarray study finding that the brains of schizophrenics showed decreased RGS-4 expression (25), and because of the location of the gene in a linkage region on chromosome 1q21-q22 of RGS-4 (26). The function of RGS proteins is to decrease the effect of G protein coupled receptor agonists. This could link with current theories on the etiology of schizophrenia relating to activity of dopamine, serotonin or metabo-tropic glutamate receptors. Other candidate genes and regions
Schizophrenia has not been convincingly associated with polymorphism in genes related to dopaminergic function, although meta-analyses have suggested a small but significant association for homozygosity at a polymorphism in DRD3 (3q13.3) (27). A modest but significant association between schizophrenia and a polymorphism in the serotonin2A receptor gene (HTR2A, 13q14-q21) was reported in a meta-analysis (28). There is a number of evidence that suggested the role for glutamatergic dysfunction in the pathogenesis or pharmacology of schizophrenia (29). Cholecystokinin (CCK, 3p22p21.3), which modulates dopaminergic neurotransmission has also been hypothesized to play a role in schizophrenia (30). In extensive review of the recent literature on molecular genetics of schizophrenia, Levinson (31) found that new genome scan project, seen in the light of previous scans, provide support for schizophrenia candidate regions on chromosomes 1q, 2q, 5q, 6p, 6q, 8p,10p,13q,15q, and 22q. Progress in identifying qualitative traits for genetic studies
There were several reports of susceptible genes or markers of vulnerability in schizophrenia-related qualitative traits such as neuropsychological measures (32), neuro-imaging measures (33), neurological signs (34) and clinical dimensions (35) Future direction
With the recently completed the haplotype structure map (‘HapMap’),
rapid decline in the cost of very high-throughput SNP genotyping and
other emerging technologies, it is likely to find more susceptible
genes and the functional significance of variations within them. The
HapMap, developed by The International HapMap Project (36) will provide
the common patterns of DNA sequence variations in the human genome,
by characterizing sequence variations, their frequencies, and correlations
between them, in DNA samples from populations with ancestry from parts
of Africa, Asia and Europe. It will help investigators across the globe
to discover the genetic factors that contribute to susceptibility to
disease, to protection against illness and to drug response. The HapMap
will provide an important shortcut to carry out candidate-gene, linkage-based
and genome-wide association studies, transforming an unfeasible strategy
to a practical one. References
© Copyright 2004 - Malaysian Journal of Medical Science
The following images related to this document are available:Photo images[mj04014t1.jpg] |
| |||||||||

{kind=link}