 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
CASE REPORT EXTRADURAL SPINAL SCHWANNOMA IN 12 YEAR OLD CHILD : A CASE REPORT Toh Charng Jeng*, Jafri Malin Abdullah*, Jain George*,John Tharakan KJ*, Sharon Casilda*, Mazira Mohamad Ghazali* Hasnan Jaafar** and Win Mar Salmah*** *Department
of Neurosciences, **Department of Pathology and ***Department of Radiology,
School of Medical Sciences, Universiti Sains Malaysia, Health Campus
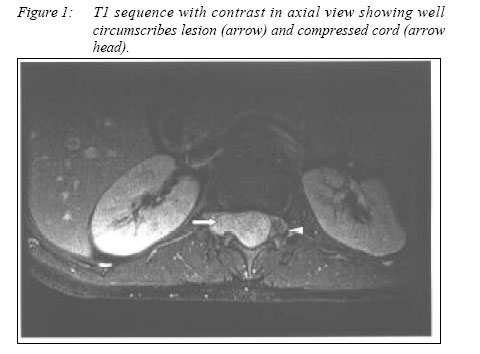
16150 Kubang Kerian, Kelantan, Malaysia Code Number: mj05022 We report a case of a 12 year old girl who presented with cord compression. Imaging studies demonstrated an extradural spinal tumour in the lower thoracic and upper lumbar levels. Histology confirmed the diagnosis of schwannoma while associated findings suggested the possibility of Neurofibromatosis Type I. Key words : Extradural spinal schwannoma, neurofibromatosis 1 Introduction Tumors of the spinal canal and its elements comprise of 5-10% of central nervous tumor in paediatric age group. Common extradural tumors in paediatric population are sarcoma, neuroblastoma, teratoma, ganglioneuroma and lymphoma. Schwannoma are rare in this age group. They usually present as intradural mass lesion and present as a dumb-bell shaped tumor and are rarely confined to the extradural space alone. Case reportA 12-year-old girl presented with subacute onset of progressive bilateral lower limb weakness. The weakness started from the right lower limb and then affected the left lower limb over a period of 2 weeks. There was presence of radicular pain at T12 dermatome of 2 months duration. She then developed hesitancy of micturation 1 week prior to admission. Past medical history revealed that she had congenital dislocation of left hip which was treated conservatively. The patient was an average student at school with an intelligent quotient (IQ) score of grade IV on Wesler Intelligence for Children. On examination, there were 4 café au lait spots (ranging from 10-30mm) but no periaxillary/inguinal freckles or subcutaneous neurofibroma. She had horizontal nystagmus, vascular anomalies of both optic discs but with normal visual acuity. Motor system examination revealed no wasting of muscles, mild spascity in both lower limbs, 0/5 muscle power in the left lower limb and 3/5 muscle power in the right lower limb. There were absent knee jerks and brisk ankle jerks with bilateral extensor plantar responses. No sensory level deficit could be detected. There was kyphoscoliosis of the spine. The upper limbs were essentially normal. Sacral reflexes were found to be normal. A formal ophthalmological assessment revealed absence of Lisch nodule. Routine haemotology and biochemical investigation were normal. Magnetic resonance imaging (MRI) of spine showed an extradural lesion extending from T12 to L1; it was isointense to hypointense in T1 and intensely enhanced with contrast (Fig 1). MRI of brain showed multiple " unidentified bright objects" in the cortex as well as near the ventricles. There was aqueductal stenosis with arrested hydrocephalus but without evidence of raised intracranial pressure. The patient had underwent surgical excision of tumour via a laminectomy. It was found during surgery that the tumor was well capsulated, firm in consistency and confined only to the extradural space. It extended from T12 to L1. The tumour was totally excised. A diagnosis of schwannoma was made on histopathological examination. The tumour was composed of spindle cells with wavy nuclei having hypocellular and hypercellular areas with focal nuclear palisading (Fig 2). The cells stained positive for S100 protein (Fig 3). Post-operatively, patient improved steadily and at 3 months she was able to walk without support. She was able to carry out her activity of daily living independently. Analysis of the NF2 gene was performed using PCR techniques. There was no mutation in this gene. DiscussionThis young girl had findings of café-au-lait spots, aqueductal stenosis with arrested hydrocephalus, mild mental subnormality, kyphoscoliosis, purely extradural schwannoma and UBOs in the certebral cortex. There was no evidence of optic glioma or vestibular schwannoma and family history was negative. These features suggested the diagnosis of neurofibromatosis type 1, although not entirely fulfilling the criteria (1,2). Diagnostic confirmations can be obtained from genetic studies. Schwannoma presenting in paediatric age group is rather rare and is especially associated with neurofibromatosis. Symptomatic intraspinal schwannoma is rarely seen in paediatric age group (3). Schwannomas are more commonly found in the intradural, extramedullary space. A study of a large series found that 68.7% of the neurinoma was intradural, 6.1% was extradural, 1.1% intra/ extradural, 5.0% had a dumbbell form, and in 1.1% cases intramedullary, and the remaining cases had neurofibromatosis (4). Schwannoma can present intraosseously and this accounted for less than 0.2% of all primary bone tumors (5). Retroperitoneal localization is rather rare (6). It is relevant to distinguish between NF1 and NF2 because of their different prognosis. Spinal nerve sheath tumors carry excellent prognosis in NF1 patients and their recurrence rate is very low. On the contrary, symptomatic neurofibromas occurring in NF2 have more severe neurological deficit, poor post-operative recovery and high recurrence rate at 10.7% at 5 years and 28.2% at 10 years respectively (7). This patient recovered well, in keeping with the diagnosis of NF1. All patients with NF1 should receive long-term follow-up for early detection and early intervention if needed in order to prevent irreversible neurological deficits especially when they start exhibiting neurological signs and symptoms. With recent technology, genetic proof is helpful in differentiating between NF1 (chromosome 17q) (8) and NF2 (chromosome 22q) (9) as done in our patient. References
© Copyright 2005 - Malaysian Journal of Medical Science The following images related to this document are available:Photo images[mj05022f3.jpg] [mj05022f2.jpg] [mj05022f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}