 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
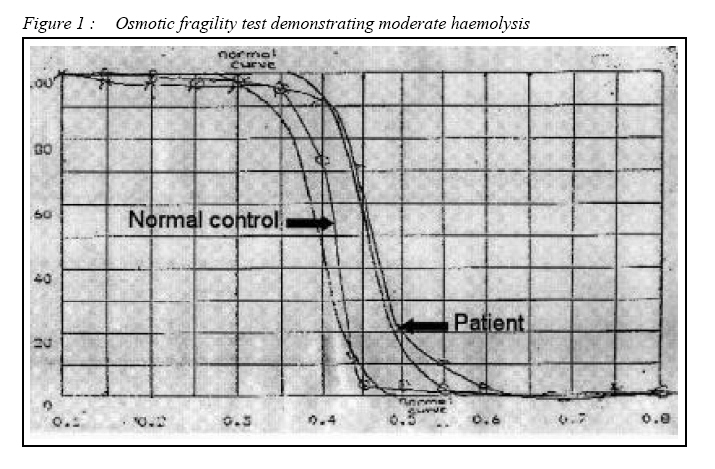
CASE REPORT Hereditary Spherocytosis in a Malay Patient with Chronic Haemolysis Muhammad Kamil Sheikh, Narazah Mohd. Yusoff, Gurjeet Kaur, Farhat Aziz Khan Advanced Medical and Dental Institute (Clinical Centre, Universiti Sains Malaysia
No. 29 Lorong Bertam Indah 4/9, Taman Bertam Indah, 13200 Kepala Batas, Penang
Malaysia. Submitted-20-02-2005, Accepted-03-12-06 Code Number: mj07026 This case report describes a 35-year-old lady who presented with generalized weakness and lethargy of two weeks duration and jaundice of more than 20 years duration. Her initial workup was suggestive of haemolysis and blood film showed a leucoerythoblastic picture with moderate microspherocytes. She was finally diagnosed as a case of hereditary spherocytosis after ruling out other possible causes of chronic haemolysis and supported by an abnormal osmotic fragility test, although family members refused for screening. Hereditory spherocytosis is uncommon in Malay population and presentation with jaundice of 20 years duration with leucoerythroblastic picture on blood film were interesting features in this case. Patient is being followed closely for need of splenectomy in near future as per severity of haemolysis and currently being managed with folic acid supplement. Key words : Hereditary spherocytosis, Jaundice, Chronic haemolysis, Leucoerythroblastic picture. Microspherocytes. Introduction Hereditary spherocytosis is described as a disorder of red cell membrane in which there is defect in red cell membrane proteins, predisposing them to haemolysis. It is characterized by presence of microspherocytes on peripheral blood film and increased osmotic fragility. Hereditary spherocytosis is commonly seen in the Caucasian population. The usual mode of inheritance for hereditary spherocytosis is autosomal dominant but autosomal recessive inheritance and sporadic cases have also been described. Most cases will have a positive family history. The clinical course of the disease is variable ranging from asymptomatic states to severe haemolysis. We report a case of a Malay lady who presented with fatigue and generalized lethargy of two weeks duration and jaundice of more than 20 years. Case Report A 35-year-old Malay lady presented with history of lethargy and fatigue for two weeks’ duration. She also complained of yellowish discolouration of the sclera since the age of 14 years which fluctuated in severity. She denied any history of fever, right hypochondriac pain, generalized itching, pale-colored stools or per rectal bleeding. Her past medical history was insignificant. However she underwent cholecystectomy in 1984 and received blood transfusion at that time. There was no history of any significant illness in the family. Physical examination revealed mild jaundice without lymphadenopathy. Her spleen was enlarged four cm below the left costal margin. Her cardiovascular examination was unremarkable so as the respiratory and central nervous system examinations. She was investigated for persistent jaundice and her blood investigation results were as follows: Full blood count - Hb. 6.2 g/dl, MCV 73 fL, MCH 23pg/cell, MCHC 32g/L, white cell count 15x109 /l and platelet count 221x109 /l. Her liver function tests revealed total protein 86g/l, albumin 47g/l, globulin 38g/l, A/G 1.2, total bilirubin 79μmol/l, direct bilirubin 13μmol/l, indirect bilirubin 66μmol/l and alkaline phosphatase(ALP) 65 iu/l. Her antinuclear antibodies (ANA) were negative and direct and indirect Coombs’ tests were also negative. HIV 1&2 (EIA), HBsAg and anti-HCV (EIA) were all negative. Peripheral blood film showed a leucoerythroblastic picture with few giant platelets and moderate microspherocytes with reticulocyte count of 4%. Ultrasound of the abdomen showed splenomegaly 15x5 cm and hepatomegaly 14cm in midclavicular line. After initial investigations and with the presence of a leucoerythroblastic picture on peripheral blood film, bone marrow aspiration and biopsy was required to exclude bone marrow infiltrative disorders. The bone marrow biopsy appearance was consistent with increased erythropoiesis. Haemoglobin electrophoresis was performed to rule out haemoglobinopathies as a cause of chronic haemolysis and the results were normal. Osmotic fragility test (Fig. 1) was performed based on the moderate number of microspherocytes on blood film and the result was a shift of curve towards the right side, indicating the presence of spherocytes. The patient was started on folic acid supplement and required blood transfusion only once. During follow up her haemoglobin level was in the range of 9g/dl and she was not transfusion dependent. However if the haemolysis worsens to a moderate or severe degree, she will be considered for splenectomy which she is not keen at the moment. Her initial presentation with acute haemolysis most likely represents concurrent illness which showed spontaneous recovery, possibly a viral illness. Discussion Hereditary spherocytosis is a heterogeneous group of disorders characterized by presence of microspherocytes which are caused by loss of membrane surface area and an abnormal osmotic fragility test. This disorder is associated with increased haemolysis, the degree of which depends on the interplay between an intact spleen and an intrinsic membrane protein defect. Hereditary spherocytosis was first described in1871 (1).The incidence in North America is 1 in 5000 (1). If osmotic fragility test is used as a screening method in blood donors it is found that extremely mild or subclinical cases do exist (2). The pathophysiology of hereditary spherocytosis involves defect in red cell membrane proteins resulting in cytoskeleton instability. Haemolysis is primarily confined to the spleen and therefore it is extravascular. Four abnormalities in red cell membranes have been described: (a) Spectrin deficiency alone (b) Combined Spectrin and Ankyrin deficiency (c) Band 3 deficiency (d) Protein 4.2 defects. A variety of mutations have been noted in genes encoding these membrane proteins. Genes responsible are localized on chromosomes 1, 2, 8, 15 and 17 for membrane proteins. Most cases are heterozygous because homozygous states are fatal. The clinical diagnosis in a typical case is usually straight forward and a family history is very important, found in nearly 75% of cases. Hereditary spherocytosis may be diagnosed at any age (3). Neonatal jaundice is common though diagnosis at this age is difficult because of the atypical appearance of peripheral film and unreliability of osmotic fragility test (4). In this patient however diagnosis was only made after patient has had jaundice and haemolysis for 20 years. Co inheritance with other haematological disorders such as β thalassaemia trait or sickle cell disease can lead to difficulty in diagnosis and give rise to variable clinical effects (5). Some neonates with hereditary spherocytosis may be transfusion dependant due to their inability to mount an erythropoeitic response in the first year of life (6). RBC(Red blood cell) morphology is distinctive yet not diagnostic. Anisocytosis is prominent, And the smaller cells are microspherocytes. Unlike the spherocytes associated with immune hemolytic disease and thermal injury, Hereditary spherocytosis spherocytes are fairly uniform in size and density. Spherocytes are characterized by a lack of central pallor, decreased Mean corpuscular volume (MCV), and increased density and increased Mean corpuscular hemoglobin concentration (MCHC) Anemia is mild to moderate. The clinical picture of hereditary spherocytosis is classified into mild, moderate or severe, this correlates with spectrin content detected in the red cells membranes in which a more severe clinical disease is associated with lower spectrin content (7, 8). The severity of the disease may be assessed based on haemoglobin level, reticulocyte count, jaundice and level of activity of patients. In a typical case diagnosis is made based on clinical history, physical examination (splenomegaly, jaundice) and laboratory data which show mild or no anemia, reticulocytosis, increased mean corpuscular hemoglobin concentration (MCHC), hyperbilirubinemia, and microspherocytes on blood film. DAT (Direct antiglobulin test) and ANA (Antinuclear antibodies) are performed to screen for major causes of autoimmune haemolysis but not all and extensive testing for autoimmune haemolysis was carried out in our case. Other supportive data include the presence of an abnormal osmotic fragility test which is performed by incubating red blood cells at body temperature whereby haemolysis of spherocytes occurs at a much lower solute concentration compared to minimal or absent lysis of normal cells. Other tests to detect membrane defects for spectrin, ankyrin, pallidin, or band 3 are possible but are not routinely available. Imaging studies may be performed to demonstrate cholelithiasis or cholecystitis. If aplastic crisis is suspected bone marrow aspiration should be performed. Management of hereditary spherocytosis includes folic acid supplements of 2.5 mg to 5 mg daily and splenectomy for patients who suffer from moderate to severe haemolysis. References
© Copyright 2007 - Malaysian Journal of Medical Science The following images related to this document are available:Photo images[mj07026f1.jpg] |
| |||||||||

{kind=link}