 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
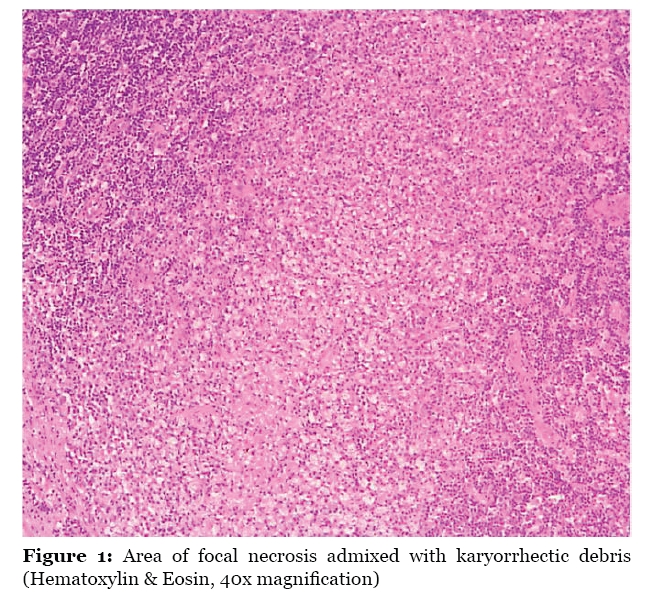
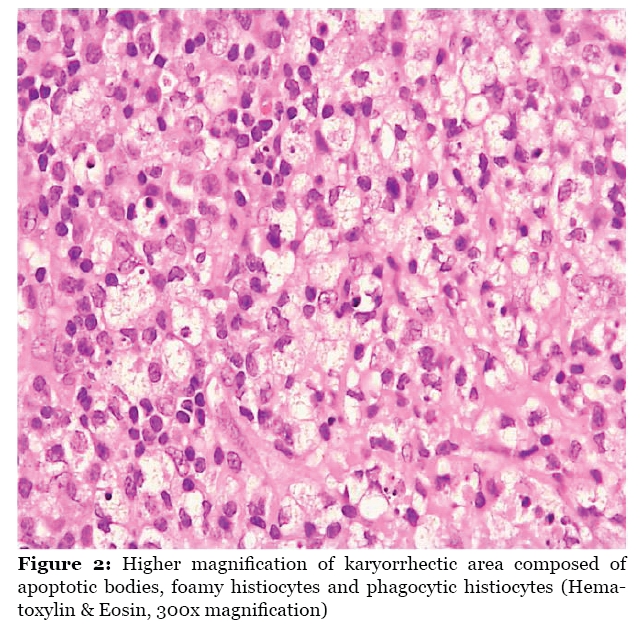
Malaysian Journal of Medical Sciences , Vol. 16, No. 4, Oct-Dec, 2009 pp. 73-76 CASE REPORT Acute Tonsillitis With Concurrent Kikuchi’s Disease as a Cause of Persistent Lymphadenopathy Halimuddin Sawali1, Primuharsa Putra Sabir Husin Athar2, Mazita Ami1, Nor Hasni Shamsudin3, Gopalan Nair4 1 Department of Otorhinolaryngology - Head & Neck Surgery, Universiti Kebangsaan Malaysia Medical Centre, Jalan Yaakob Latif, Bandar Tun Razak, 56000 Kuala Lumpur, Malaysia Submitted: 18 Feb 2009 Code Number: mj09037 Abstract We present a young adult female with symptoms of acute tonsillitis and tender cervical lymphadenopathy. Despite a full course of oral antibiotics, she had persistent left lower cervical lymphadenopathy measuring 2.0 x 1.5 cm at 2 weeks post-treatment. Rigid and flexible scope examinations did not reveal any abnormalities in the nasopharynx, oropharynx or hypopharynx. Tuberculosis tests were negative and blood index results were normal. Fine needle aspiration cytology revealed a non-specific granulomatous inflammatory process. Excisional lymph node biopsy was performed, and the patient was diagnosed as having Kikuchi’s Disease (KD). We would like to highlight the diagnostic challenges in detecting this condition and the importance of differentiating KD from tuberculosis and malignant lymphoma, the latter of which requires aggressive treatment. Keywords: histiocytic necrotizing lymphadenitis, Kikuchi disease, Kikuchi-Fujimoto disease, medical sciences Introduction Kikuchi-Fujimoto disease (histiocytic necrotizing lymphadenitis) is a benign, self-limited disease of unknown cause that often manifests with persistently enlarged cervical lymph nodes that are unresponsive to antibiotic therapy (1). The disease was first identified by Kikuchi in 1972; this condition is primarily seen in young adult women, frequently of Asian origin, and is becoming increasingly recognized worldwide (2). The incidence of Kikuchi’s disease (KD) ranges from 3% to 9% of all biopsy cases of neck lymph nodes (3). Approximately 30% of KD patients are mistakenly thought to have malignant lymphoma. Therefore, it is important for clinicians and pathologists to be aware of this benign, self-limiting entity and to differentiate it from non-Hodgkin’s lymphoma, systemic lupus erythematosus (SLE), and carcinoma. Case Report A 22-year-old female presented with a five day history of sore throat and fever with bilateral cervical lymph lymphadenopathy. Examination revealed a high fever of 38.5 °C, mildly enlarged inflamed tonsils and bilateral tender cervical lymphadenopathy. She was treated with a course of antibiotics for a week. She recovered well except for a persistent mobile left lower cervical lymph node measuring 2.0 x 1.5 cm, which did not regress even at 2 weeks post-treatment. There were neither other lymphadenopathies nor hepatosplenomegaly. Investigations for tuberculosis (TB) were negative. White cell counts were normal. Fine needle aspiration cytology (FNAC) of the lymph node was suggestive of an acute granulomatous inflammatory process. Rigid nasal endoscopy and flexible nasopharyngolaryngoscopy did not reveal any abnormality in the nasopharynx or hypopharynx. An excisional lymph node biopsy was performed and histopathological examination reported KD (Figure 1 and Figure 2). Further blood investigations for connective tissue screening (antinuclear antibody, rheumatoid factor) were negative. Levels of complements C3 and C4 were also normal. The patient recovered well after excisional biopsy of the lymph node. It has been 8 months since the surgery and there has been no recurrence. Discussion Diagnosis of KD is easily missed and readily underdiagnosed. It is more common than previously thought (4). Menasce et al. (5) performed a study in UK that reported that KD is still a poorly recognized entity and is frequently confused with malignant lymphoma. They re-examined 27 lymph node biopsies from 25 patients who were diagnosed with KD. Only three cases showed that the initial pathological diagnosis was Kikuchi disease; the most common suggested diagnosis was nonHodgkin’s lymphoma (5). Diagnosis of KD is rarely made as a provisional diagnosis until all other possible causes of cervical lymphadenopathy have been ruled out, especially in South East Asia region, where tuberculosis is endemic. In most cases, persistence of lymphadenopathy after completion of antibiotics will raise suspicion of this disease, which can only be confirmed by histopathological examination. In this case report, KD is an incidental finding. Even though both acute tonsillitis and KD can present with cervical lymphadenitis, the presence of sore throat with inflamed tonsils favour the diagnosis of acute tonsillitis during the initial presentation. Acute tonsillitis would usually respond to a course of antibiotics, which is not the case for KD. Therefore, in this case, we postulate that KD occurred concurrently with tonsillitis. Histologically, KD must be differentiated from non-Hodgkin’s lymphoma, SLE and carcinoma. It is very important to differentiate between these diseases because the treatment regimen and prognosis are specific to each condition. Based on the histology of Kikuchi disease, it can be further divided into four subtypes: lymphohistiocytic type, phagocytic type, necrotic type and foamy cell type. Kikuchi et al. (2004) reported that lymphohistiocytic and phagocytic are the more common types, which constitute more than 80% of cases; the other two types are occasionally encountered. The symptom of high-grade fever greater than 39oC is commonly seen in cases of the necrotic type (6). The typical pathology of KD permits differentiation from lymphoma, SLE, and infective lymphadenopathies (7). The classic morphology of KD includes patchy lymph node involvement centred in paracortical areas, extensive fibrinoid necrosis, absence of granulomatous reaction, and foamy histiocytes at the margins of the necrotic areas. Plasma cells are rarely noted and abundant predominantly extracellular apoptosis debris is present (8). Typical features of infiltration by blasts and histiocytes with extensive necrosis or karyorrhexis are shared with other conditions, such as malignant lymphoma, tuberculosis, and SLE (9). The features that distinguish lymphoma from KD include the presence of cytologically bland histiocytes, absence of a prominent “starrysky” appearance, presence of predominantly extracellular “nuclear dust,” absence of neutrophils, eosinophils, and plasma cells in areas of necrosis and a predominance of T immunoblasts. Reed-Stenberg cells are also not a feature of KD. Pertaining to immunohistochemical studies, CD68 highlights crescentic histiocytes and plasmacytoid monocytes, unlike non-Hodgkin lymphoma, in which it shows sheets of B or T cells. In KD, immunoblasts are negative for CD15 and positive for LCA (CD45) (8). Histologic features of SLE lymphadenopathy may not be distinguishable on a morphologic or immunophenotypic basis alone. Presence of numerous plasma cells, DNA deposition on vessel walls and extensive areas of acellular coagulative necrosis devoid of karyorrhectic material would favour the diagnosis of SLE (8). Even though KD is rarely associated with SLE, but its diagnosis can precede, postdate or coincide with SLE because lymphadenopathy is a common presentation in both conditions and histologically may be indistinguishable between them (10). Although the aetiology of KD is unknown at present, it has long been speculated to be of autoimmune or infectious origin (2). Possible links between KD and systemic lupus erythematosus and a nonspecific hyperimmune reaction to a variety of infections, chemical, physical, and neoplastic agents has also been proposed for its pathogenesis. As in this case, the young female patient had presented with typical symptoms of an upper respiratory tract infection that resolved with oral antibiotics; however, the left lower cervical lymph node remained persistent. Further investigations, which included FNAC of the node, TB work-up and flexible scope examination of the nasopharynx and hypopharynx did not lead to any conclusion. Excision biopsy of the node had to be carried out to establish a KD diagnosis for this patient. KD is a self-limiting disease; the lymphadenopathy slowly resolves after a few months. Prognosis is excellent, but about 3% of patients show recurrence of the disease within several years; one study reports this number to be 2% to 5% of cases (3). Some authors advocate the use of corticosteroids and reported faster healing responses in terms of lymphadenopathy disappearance. Meanwhile, other conditions that share similar clinical presentations, such as malignant lymphoma, SLE and tuberculosis, have specific, aggressive and prolonged treatments. Conclusion KD, although rare, has become a more recognized condition since it has been reported not only in Asia, but around the world as well. This diagnosis should be seriously considered, especially in patients presenting with fever and persistent cervical lymphadenopathy. Including this disease as a differential diagnosis when histopathological examination is requested will improve early detection. By identifying KD early, expensive and extensive investigations that may lead to unnecessary treatments can be avoided. Author’s contributions
References
© Copyright 2009 - Malaysian Journal of Medical Science The following images related to this document are available:Photo images[mj09037f2.jpg] [mj09037f1.jpg] [mj09037f21.jpg] |
| |||||||||

{kind=link}
{kind=link}