 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
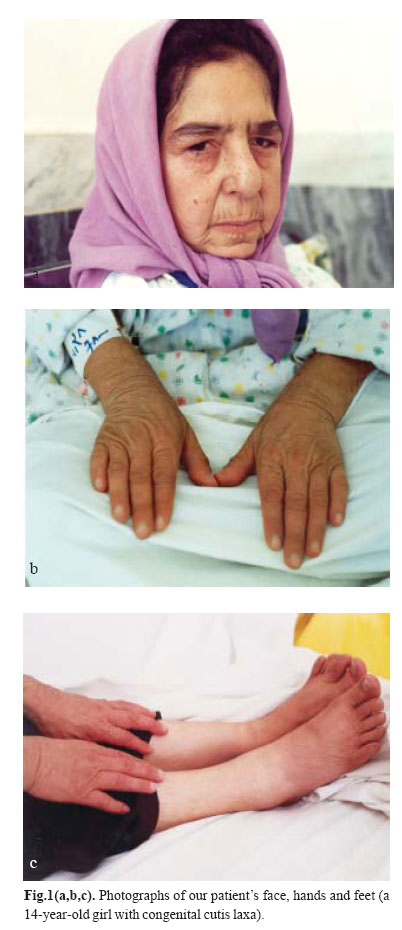
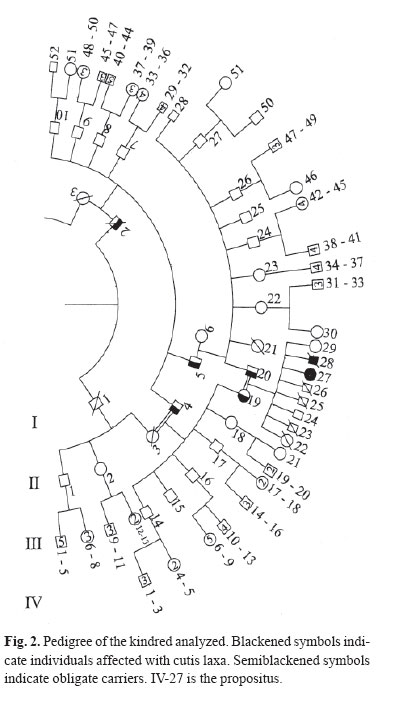
Medical Journal of the Islamic Republic of Iran , Vol. 18, No. 1, May, 2004, pp. 87-89 Case Reports PRESENTATION OF A PEDIGREE OF AN IRANIAN FAMILY WITH TWO MEMBERS WITH CUTIS LAXAAUTOSOMAL RECESSIVE TYPE I H. POUR-JAFARI*, Ph.D., M.G., AND A. SARIHI,** Ph.D.From the Departments of *Genetics and **Physiology, Medical School, Hamadan University of Medical Sciences, Hamadan, I.R. Iran. ABSTRACTCongenital cutis laxa is an exceptional condition. No large scale pedigree has been reported from Iran. We report a family with 106 members with two members affected with cutis laxa. Our cases were two patients (male and female) with pre- and postnatal growth retardation, cutis laxa, characteristic facies and other manifestations which proved that they were affected with cutis laxa. Their family history was studied and a large pedigree was drawn up. Based on the findings in their pedigree pattern, in addition to clinical and pathological studies, one can say that cutis laxa in this family is autosomal recessive. We also showed obligate carrier members in the family. Recent studies have shown that cutis laxa is a heterogeneous group of conditions both clinically and genetically.Autosomal dominant, autosomal recessive, X-linked and also acquired forms have been reported. Our study indicates that our case is an autosomal recessive type I. We discussed the pedigree that covers five generations. Keywords: Abnormalities, Cutis laxa, Hereditary diseases. INTRODUCTION Goltz et al. described affected brothers and suggested recessiveness because of other reported instances of affected sibs as well as parental consanguinity.1 Different studies have shown that cutis laxa is a heterogeneous group of conditions both clinically and genetically. Autosomal dominant,2 autosomal recessive,3 Xlinked, 4 and also acquired forms5 have been reported. Our research group recently worked on a case of cutis laxa that is alive, and her dead brother who was affected too. Her hanging skin, giving the appearance of premature aging and her other clinical, para-clinical and genetic features were studied. Based on clinical manifestations and laboratory findings, we found that our cases are affected with CCL (congenital cutis laxa type I).6 We studied their family through different approaches. The aim of the present study is to determine the mode of transmission of the mutant gene through our case family. CASE REPORT Our primary case was a 14-year-old Iranian girl affected with congenital cutis laxa (Fig. 1). We studied her clinical manifestations, laboratory findings and also her family history. She had pre- and post-natal growth retardation, cutis laxa, characteristic facies, cardiac failure caused by gross pulmonary emphysema, chest infections, lax vocal cords and some other clinical manifestations, along with pathological findings which totally proved that she was affected with congenital cutis laxa type I. Indeed, complete clinical and family studies and skin biopsy with standard histology, orceine staining and histomorphometric analysis of the collagen and elastic fibers of the dermis were performed to prove that our case was a case of congenital cutis laxa. Her family history showed that she had another sib that based on his hospital information was affected with similar disease. He had died at the age of two years. We drew up their pedigree containing four generations and 106 members. Fig. 2 shows the pedigree of the kindred analyzed. Based on the following observations, it seems that the pedigree pattern of cutis laxa in the subject family is autosomal recessive: a) Presence of affected sibs. b) Parents of propositus had normal phenotypes. c) Parental consanguinity (first cousins). DISCUSSIONAs results showed the mode of transmission of cutis laxa type I in our subject family was autosomal recessive. If this suggestion is correct, we can assume that: a) The parents of propositus are obligate carriers; b) The future progenies are at 25 percent risk for being affected; c) Normal progenies are at about 66 percent risk to be a carrier; d) One of these two individuals, II-3 or II-4 is an obligate carrier, and the other one is a noncarrier; e) Based on the authors' best knowledge, II-6 is not a carrier, because in her family history (three generations), despite some consanguineous marriages there was no person with CCL. f) Probably I-2 is the first one that was a carrier or may have the de novo mutant gene. There are several reports that show the mode of inheritance of the disease to be autosomal recessive. Rybojad et al. reported inheritance of cutis laxa in a group containing five children with cutis laxa.7 He showed that cutis laxa is an autosomal recessive disorder in four cases out of five. Khakoo et al. reported two cases of congenital cutis laxa with autosomal recessive inheritance.8 Other researchers also presented similar suggestions;9,10 but some investigators reported that their observations have shown that CCL is transmitted as an autosomal dominant trait .2, 11, 12 It seems the disease which is named under the entity congenital cutis laxa, is a heterogenous abnormality. Review of clinical manifestations and paraclinical findings of cases that are reported in recent years may clarify the different nature of the genes causing the group of abnormalities which are called cutis laxa in general. ACKNOWLEDGEMENTWe hereby would like to thank Professor T. Brown and Mr. R. W. Sorfleet for their kind review of the article and in suggesting some corrections and Hamadan University of Medical Sciences, Research Affairs, for financial support of this research. REFERENCES
Copyright 2004 -Medical Journal of the Islamic Republic of Iran The following images related to this document are available:Photo images[mr04015f2.jpg] [mr04015f1.jpg] |
| |||||||||
{kind=link}
{kind=link}