 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
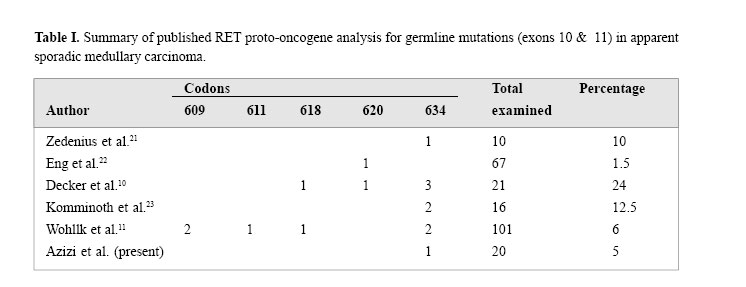
Medical Journal of the Islamic Republic of Iran , Vol. 18, No. 2, August, 2004, pp. 95-99 Original Articles: Clinical Science THE MUTATIONS OF RET PROTO-ONCOGENE INMEDULLARYTHYROID CARCINOMAS IN IRAN IRAJ NABIPOUR, M.D., FATEMEH HAJI-GHASEMI, M.Sc.,SHAHRIAR KIAI, M.Sc., REZA BARADAR-JALILI, M.D., ANDFEREIDOUN AZIZI, M.D. From the Endocrine Research Center, Shaheed Beheshti University of Medical Sciences, Tehran, thePersian Gulf Health Research Center, Bushehr University of Medical Sciences, Bushehr and the EndocrineResearch Center, Tehran University of Medical Sciences, Tehran, Iran. Code Number: mr04017 ABSTRACTMedullary thyroid carcinoma (MTC) occurs both sporadically and in the autoso-mal dominantly inherited multiple endocrine neoplasia (MEN) type 2 syndromes. The distinction between true sporadic MTC and a new mutation familial case is important for future clinical management of both the patient and family.The susceptibility gene for hereditary MTC is the RET proto-oncogene. DNA analysis for germline mutations of the RET proto-oncogene was performed in a series of 24 patients with MTC [appar-ently sporadic MTC (20 cases), familial MTC (2 cases), MEN 2A (one case) and MEN 2B (one case)] to determine whether they were true sporadic cases or heredi-tary forms. Genomic DNA was amplified using polymerase chain reaction (PCR) and oligonucleotide primers for exons 10 & 11. The PCR products were examined by restriction enzymes analysis to detect the mutations. One of the 20 patients with appar-ent sporadic MTC had exon 10 mutation (Cys-620 Arg); and exon 11 mutation (Cys-634 Trp) was also found in the index case with MEN 2A. No mutation was detected in the other patients. Three of six evaluated members of the MEN 2A patient had the same mutation. We conclude that routine application of RET proto-oncogene testing should be included in all cases of apparent sporadic MTC. INTRODUCTION An estimated 20-25% of medullary thyroid carcinomas (MTC) appear in the context of inherited disease, i.e. in the multiple endocrine neoplasia type 2 (MEN 2) syndromes and the familial form of MTC, FMTC.1,2 The RET proto-oncogene is expressed in cells of neuronal and neuroepithelial origin and encodes a receptor tyrosine kinase. The first 10.5 exons encode the extracellular region, which includes a cadherin-like and a cysteine- rich domain.3 The intracellular tyrosine kinase domains and the C-terminal tail are encoded by the remaining exons. The highly conserved cysteine domains are pivotal in maintaining the secondary and tertiary structures of the RET extracellular domain. Mutations in these cysteine domains, in codons 609, 611, 618 and 620 of exon 10, and of codon 634 of exon 11, enhance the ligandindependent dimerization and cross-phosphorylation, thus allowing constitutive protein tyrosine kinase activity in the absence of the ligand.4 In 1993, germline point mutations in RET were identified that segregated with the disease phenotype in MEN 2 and FMTC families.5 About 98% of MEN 2A and 88% of MTC families have germline missense point mutations in the cysteine codons of the extracellular and transmembrane domains of the RET receptor molecule (encoded by exons 10 and 11). Germline mutations also occur in the intracellular domain of RET in FMTC: in exon 13 (codons 768, 790, and 791), in exon 14 (codon 804 and 844), and in exon 15 (codon 891).6 Genetic testing for germline mutations in the RET proto-oncogene has become available and today forms the basis for DNA-based screening procedures. Molecu-lar biology now affords an early identification of carriers of RET proto-oncogene germline mutations who are sus-ceptible to develop MTC later in life. In these patients, early prophylactic thyroidectomy must be envisaged to ensure definite cure.7 This RET mutation analysis is very useful as a means of excluding genetic susceptibility to the development of MTC and therefore of assuring unaffected family members that further screening with biochemical tests can safely be abandoned.8 Based on the relative frequency of different MEN 2/ FMTC phenotypes and the association with specific RET defects, a strategy for priorization of mutation detection can be proposed. Initially, the mutation detection should focus on exon 11 (codon 634), followed in order by exon 10 (609, 611, 618, 620) and exon 16 (codon 918). This strategy will identify a RET mutation in most cases of hereditary MTC. The cases in which no mutation is found should be considered for sequencing of exons 13, 14, and 15 to search for rare mutations.9 Unfortunately, no mutation analysis has been done for Iranian patients with MTC. Therefore, in this study, 24 subjects affected by MTC, without regard to family history or the presence of C-cell hyperplasia who were followed up in university hospitals in Tehran/Iran that focus on the management of MTC were examined to de-tect the possibility of the presence of RET proto-oncogene mutations in exons 10 and 11. MATERIAL AND METHODSPatients The study population included 24 patients with MTC who were operated in different university hospitals and institutions of Tehran and had clinical follow-ups within the preceeding five years. We examined the consecutive cases of MTC in these hospitals and institutions, with-out regard to family history or the presence of C-cell hyperplasia. The studied subjects included 20 cases with apparently sporadic MTC, 2 patients with familial MTC, MEN 2A (one case) and MEN 2B (one case) subjects according to the International RET Mutation Consor-tium.8 The mean age of the patients (11 males & 13 females) was 42.5 years. The diagnosis of MEN 2A was based on the presence of MTC and pheochromocytoma in a 65 year old male. One patient with MEN 2B was a 10 year old girl. She had MTC, mucosal neuromas and a marfanoid habitus. Two patients with FMTC were re-lated subjects, whereas clinical and laboratory data ex-cluded the presence of pheochromocytoma, hyperpar-athyroidism and the other features of MEN 2. The diag-nosis of apparently sporadic MTC was based on the absence of familial history of disease and the clinical and laboratory features of MEN 2. All patients with MTC were treated by at least a standard thyroidectomy in conjunction with a standard systematic cervico-central lymph node dissection. Direct mutation analysisGenomic DNA was extracted from peripheral leukocytes and was amplified using PCR and oligonucleotide primers for exon 10, (10F 5’ GCGCCCCAGGAGGCTGAGTG3’ and 10R 5’ CGTGGTGGTCCCGGCCGCC3’) and exon 11, (11AF5’ CCTCTGCGGTGCCAAGCCTC3’ and 11AR 5’ CACCGGAAGAGGAGTAGCTG3’). Exons 10 & 11 were amplified as described previously by Wells and et al.11 The reactions were carried out in an automatic thermocycler (OmniGene & Hybaid Co.). Individual aliquots of PCR products from exon 10 subsequently were incubated with one of six restriction enzymes Nla I, Mbo II, BstU I , Rsa I , Taq I [New England Biolabs, Beverly, MA] and Cfo I [Roche Molecular Biochemicals] according to conditions specified by the manufacturers. Similarly, aliquots from exon 11 reaction mixtures were incubated with one of four restriction enzymes Cfo I, Rsa I, Hae III, and Dde I . Reaction products were sepa-rated by electrophoresis through 10% nondenaturing polyacrylamide gels and detected by ultraviolet visual-ization after ethidium bromide (EtBr) staining. RESULTSGermline RET mutations in exons 10 and 11 were iden-tified in 2 of 24 patients with MTC. One of them was a patient with apparent sporadic MTC who had a muta-tion in exon 10 and the other patient had MEN 2A with a mutation in exon 11. No patient with FMTC had a germline mutation. Exon 10 mutationA codon 620 (Cys 620 Arg) [TGC>CGC] was found in a 75 year old man with apparent sporadic MTC. The pa-tient presented with hoarseness and neck mass who was treated by total thyroidectomy and lymph node dis-section four years ago. Mutation analysis was done when he was admitted in one of the university hospitals with metastatic manifestations of the lungs. A careful family history failed to uncover a history of MTC. The patient had expired in the I.C.U. when the result of RET proto-oncogene analysis was provided, and his family could not be found due to change of address. Exon 11 mutationA codon 634 (Cyst634 Trp) [TGC>TGG] mutation was identified in the 65 year old index case. He was a known case of MEN 2A since 21 years ago when adrena-lectomy had been done preceeding thyroidectomy. The propositu’s sister died with MTC and pheochromocy-toma at age 50. The index case has nine children. Four (3 females & one male) of his children had been treated by total thyroidectomy and adrenalectomy (except a 28 year old daughter who had only MTC). Mutation analysis of the affected propositu’s children revealed a codon 634 mutation in exon 11; his unaffected 41 year old daughter had no mutation. His affected 39 year old son has three (2, 9, and 12 year old) males and a 4 year old female child. A codon 634 mutation in exon 11 was found in the 12 year old male of this examined second generation. We recommended thyroidectomy for this gene carrier. DISCUSSIONIn our study, of twenty cases with apparent medul-lary carcinoma, we identified germline mutation in exon 10 in a patient by analysis of RET proto-oncogene, indi-cating the presence of a sporadic misclassified familial disease. The identification of MTC in a single patient with no family history of MTC or MEN 2A poses a dilemma for the clinician. There exists the possibility that the affected individual could be the proband for a new kindred or may represent a de novo mutation that could be transmitted to children. Four series examined the frequency of occult or de novo germline RET mutations in MTC cases. One series examined consecutive cases of MTC without regard to family history or the presence of C-cell hyperplasia and found a 25% occult RET mutation frequency.12 The other three series examined cases of apparently sporadic MTC without known family history and without features sug-gestive of MEN 2. The occult or de novo mutation fre-quency among these apparently sporadic cases ranged from 2.5% to 7%.13,14 Table I compares our study with other published RET proto-oncogene analysis for germline mutations in ap-parent sporadic medullary carcinoma. We can envision two ways in which RET proto-oncogene mutational in-formation will be helpful in the management of cases of apparent sporadic MTC. The most obvious is the identi-fication of previously unrecognized examples of MTC or MEN 2A where other family members are at risk for de-velopment of disease. The identification of a specific mutation provides a straightforward method for screen-ing first degree relatives. Another benefit, perhaps even more significant for first degree relatives of an individual with sporadic MTC, is the exclusion of hereditary disease.Therefore, RET proto-oncogene analyses should be performed on all patients with apparent spo-radic MTC.13 Familial medullary thyroid carcinoma (FMTC) con-sists of pedigrees in which MTC is transmitted as an autosomal dominant trait without other manifestations of MEN 2. Until recently the FMTC phenotype was re-lated in 80-96% of cases to RET mutations located in exon 10 (mainly at codons 620 and 618) and in exon 11 (codon 634) (15 & 16). Gemline mutations also occur in the intracellular domain of RET in FMTC: in exon 13 (codons 768, 790, and 791), in exon 14 (codon 804 and 844), and in exon 15 (codon 891).17,18 In a large series of patients, 148 patients from 47 FMTC only families, noncysteine RET mutations were found in 59.5% of these families. This study showed that FMTC with noncysteine RET mutations are not infrequent and are overrepre-sented in presumed sporadic MTC, suggesting that RET analysis should routinely be extended to exons 13, 14 and 15.19 We did not detect mutations in 2 FMTC index cases by RET proto-oncogene analysis in exons 10 and 11. When the search for mutations is extended to other RET exons, some rare mutations in exons 13-15 may be observed. Some 93-98% of MEN 2A families have germline missense mutations in the extracellular domain of the RET proto-oncogene in exon 10 (codons 609, 618, 620) or in exon 11 (codon 634). The most frequent mutation in MEN 2A is located at codon 634.16 A codon 634 mutation was also identified in the 65 year old index case with MEN 2A in our study. The same mutation was present in a 12 year old son of the affected 39 year old male of the index case with MEN 2A. Consensus was reached at the MEN97 Workshop that the decision to perform thyroidectomy in MEN 2 should be based predominately on the result of RET mutation testing, rather than on CT testing.20 Children with any RET codon 611, 618, 620, or 634 muta-tion are classified as level two or as having a high risk for MTC and should have thyroidectomy performed be-fore the age of 5 years.21 Therefore, we recommended thyroidectomy for this 12 year old gene carrier. MEN 2B syndrome consists of MTC, pheochromocytoma, mucosal neuromas, ganglioneuromatosis of the gut, and marfanoid habitus.22 Children with MEN 2B and/ or RET codon 883, 918, or 922 mutation are classified as level 3 or as having the highest risk from aggressive MTC and should have thyroidectomy within the first 6 months and preferably within the first month of life.21 A single missense mutation in the intracellular tyrosine ki-nase domain, in codon 918 in exon 16, occurs in 95% of patients with MEN 2B.6 Unfortunately, we did not ex-tend analysis of RET proto-oncogene mutations in exon 16 (codon 918) in our patient with MEN 2B. A few germline RET proto-oncogene mutational analysis had been done in Asia.23-25 But as our study, only exons 11 & 10 had been evaluated in these studies. Large-scale analysis of mutations in RET exon 16 in sporadic medullary thyroid carcinomas in Japan was performed.26 Our case series of medullary carcinoma is unique in number in the west of Asia, therefore we will extend germline RET mutation analysis of our patients to exons 16, 14, and 15. Because of the critical clinical implication of finding a RET mutation, sequence analysis will also be performed directly for the exons. REFERENCES
Copyright 2004 -Medical Journal of the Islamic Republic of Iran The following images related to this document are available:Photo images[mr04017t1.jpg] |
| |||||||||
{kind=link}