 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
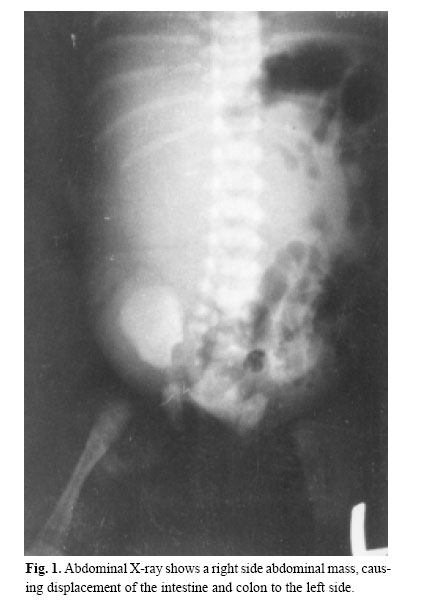
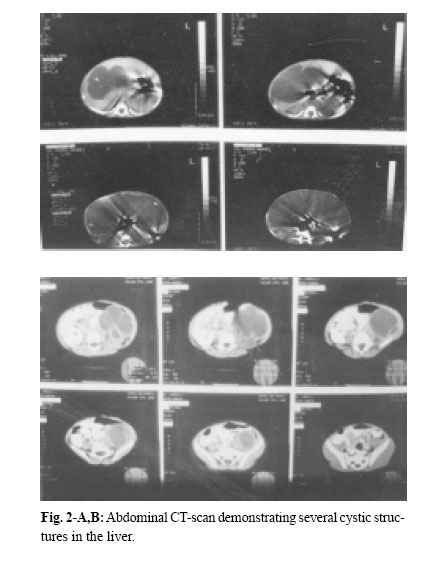
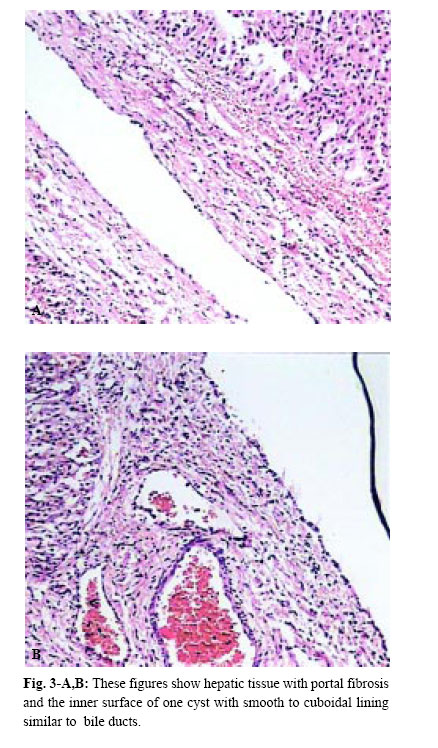
Medical Journal of the Islamic Republic of Iran , Vol. 18, No. 2, August, 2004, pp. 185-189 A NEONATAL PRESENTATION OF CAROLI’S DISEASE WITH SEVERE ABDOMINAL DISTENTION AS A PRESENTING SYMPTOM From The Department of Pediatrics and Pediatric Surgery, Ghaem Medical Center, Mashhad University of Medical Sciences, Mashhad,I.R. Iran *Associate Professor of Pediatric Surgery Code Number: mr04034 In this article , the authors introduce a case of Caroli’s Disease (CD) according to the results of ultrasonography (U.S), abdominal computed tomography (CT scan) and hepatic biopsy that has been manifested in the neonatal period. In the routine examination of the neonate, abdominal protrusion was noticed. Then milk intoterance, inability of meconium passage and vomiting developed, during workup, a diagnosis of CD was confirmed. Although CD is rare in the neonatal period, it should be considered in the differential diagnosis of abdominal protrusion. Keywords: Caroli’s Disease, Caroli syndrome, Intrahepatic bile duct dilation, Abdominal protrusion, Neo-nate. Caroli’s disease is a rare congenital abnormality of the intrahepatic bile ducts, characterized by multifocal cystic or saccular nonobstructive dilatations of the intrahepatic bile ducts.1 This disease is considered to be an autosomal recessive inherited condition, more than 200 cases of CD have been reported in the literature.2 The clinical course can be asymptomatic for the first 15-20 years or symptoms can occur infrequently throughout the patient’s life. Repeated episodes of bacterial cholangitis and hepatolithiasis develop in 50% of patients due to bile stagnation. Secondary biliary cirrhosis usually follows years later. A neonatal presentation of CD with severe cardiac and progressive renal pathology has been described.3 Here we present a case of CD in a one day old girl with abdominal protrusion as a chief presentation. According to our available data, this is the first case of CD in the neonatal period with such a presentation. On December 16, 2000 at one of Mashhad’s Hospitals a female neonate was born by a 22 year-old mother, in her first pregnancy by normal vaginal delivery. The newborn’s weight was 3500 g head circumference and length were 34 cm and 54cm respectively and Apgar score of 8-8. The blood group of the mother was B+ (neonate A+). Just after birth her abdominal distention could be seen. Her mother had had no disease or medications during pregnancy, and had not received any special prenatal cares. Amniotic fluidt volume was normal and appeared clear but because the mother had premature rupture of amniotic membranes (20 hours before labor), she was hospitalized and antibiotic therapy begun for her. After labor, obstetric management was done because of severe vaginal hemorrhage. Because of abdominal distention, poor feeding and vomiting the neonate was transferred to NICU for care and workup. In physical examination, the neonatal reflexes were normal and during the first 24 hours of life. The urine output was normal, but had no meconium passage. Auscultation of the heart and lungs was all right. The abdomen was soft but distended and a large mass was palpated in the right hemiabdomen. The extremities and genital system were all right but the anus was anterior. Laboratory data& imaging study results were as follows: Chest X-ray showed elevation of the right diaphragm. In the supine abdominal X-ray, an opacity on the right side which had pushed intestinal gases to the left side of the abdominal cavity was observed (Fig. 1). The abdominal ultrasonography revealed a large multicystic mass with septation that had attachment to the inferior border of the right lobe of the liver. The spleen, pancreas and kidneys except for a mild dilatation in the collecting system of the left kidney, were normal. The abdominal CT scan showed normal kidneys and spleen but a large cystic lesion that had occupied most of the abdominal cavity (Fig. 2-A,B). On the second day of life, laparotomy was performed and revealed a large cystic mass that had occupied most parts of the abdominal cavity including both lobes of the liver. Other intraabdomianl organs were normal. Partial resection of the mass, including lower borders of the right and the left lobe of the liver was performed. Macroscopic description: a cystic tissue mass, with hyperemic and gangrenous surfaces, a smooth inferior surface and elastic consistency,11*6.5*6 cm in diameters combine with a small margin of the hepatic tissue was seen. In the dissection, the mass contained a green biliary substance. Microscopic description (by light microscope): the mass was a part of the liver that was filled with multiple small cystic lesions with lining similar to the bile ducts and marginal and port space fibrosis (Fig. 3-A,B). According to the above descriptions, the diagnosis is compatible with Caroli’s disease. Postoperative period was complicated by icter, gastrointestinal and pulmonary hemorrhage and liver failure (in spite of supportive cares in NICU), finally, on December 22, 2000 (six days after birth), the neonate expired. Caroli’s disease is a rare condition and was first described by Caroli and Carcosin in 19581 and classified later as type V choledochal cysts, or as type IVA if they are associated with an extrahepatic choledochal cyst as they are in 21% of cases.4,5 Congenital, sacular dilatation may affect several segments of the intrahepatic bile ducts. The dilated ducts are lined by cuboidal epithelium and are in continuity with the main duct system, which is usually normal. Caroli actually described two variants: 1-Caroli’s disease characterized by ectasias of the intrahepatic bile ducts without other abnormalities, and 2-Caroli syndrome, in which congenital ductal dilation is associated with features of congenital hepatic fibrosis and the renal lesion of autosomal recessive polycystic renal disease.6 Caroli syndrome is more common, but both varieties may occur in the same family and are inherited in an autosomal recessive fashion. Loss of the distal 3p chromosome and gain of the 8q chromosome is of pathogenic importance.2 The pathogenesis of CD seems to involve total or partial arrest of remodeling of the ductal plate of the larger intrahepatic bile ducts.7 Caroli’s disease may be confined to one segment or lobe but is usually diffuse.8 This problem also appears to be more common in Asia. The usual main symptoms of CD are fever and abdominal pain, with or without jaundice. Cholangitis is the main presenting symptoms in 64% of patients, portal hypertension in 22%, recurrent abdominal pain in the right upper quadrant in 18% and pyelitis, systemic hypertension and hematuria in 2%.7 Our patient is the first case of CD that manifested in the first birth day with abdominal protrusion. The available imaging techniques for the diagnosis of CD are ultarsonography, percutaneous transhepatic cholangiography, radionuclide scintigraphy, computed tomography (CT), endoscopic retrograde cholangiopancreatography (ERCP) (The diagnosis was possible because of the availability of 7.5 mm pediatric duodenoscope which allows ERCP to be performed safely even in very small infants)12 and magnetic resonance cholangiopancreatography (MRCP).13 The sonographic appearance in CD is intrahepatic cystic anechoic areas in which fibrovascular bundles, stones and linear bridging or septum may present, the fibrovascular bundles are composed of portal vein& hepatic arteries, which can be demonstrated by Doppler blood flow.14,15 CT images show intrahepatic cystic dilatation of the biliary tree, plus bulges in the wall of the saculus, septa and fibrovascular bundles. ERCP shows communication between the sacculi and bile ducts. These findings can differentiate C.D. from liver cysts. Complications from CD are bacterial cholangitis (64%), hepatic abscess, hepatolithiasis (34%), gallbladder stones (21%) and portal hypertension (22%). Dysplasia of the biliary epithelium in CD is considered to be a premalignant lesion. It is reported that 7% of CD cases may progress to cholangiocarcinoma.2 The major aim of medical treatment for CD is to reduce the morbidity and mortality of recurrent cholangitis, hepatic abscess and biliary obstruction. There are 3 situations that require special attention in term of management, monolobular Caroli’s, diffuse Caroli’s and antenatally diagnosed Caroli’s. For monolobular, hepatic resection is the preferred treatment because this gives complete and long lasting resolution of symptoms.16 The diffuse form has several treatment options: cholangitis is treated with antibiotics and drainage. ERCP with sphincterotomy, stone extraction and then stenting, external and internal biliary drainage operation have been used to remove the biliary obstruction. In some instances, ursodeoxycholic acid has been used to dissolve intrahepatic stones When complications such as biliary cirrhosis, severe portal hypertension or cholangiocarcinoma arise, liver transplant then is the only option.17 In summary by revisiting the related articles, one can see that CD is very rare in the neonatal period. In April 1997 a neonatal presentation of C.D. with severe cardiac and progressive renal pathology is described.3 In October 1998 another group researchers reported three neonates with anuria, hepatomegaly and Jaundice.18 Also in Sep. 2000 Ioama et al. reported an infant who had a multiloculated cystic lesion located in segment IV of the liver consistent with CD, diagnosed by routine prenatal ultrasound at 25 weeks gestation and confirmed by HIDA scan and computed tomography soon after birth and gradual regression of the cysts noted by serial sonograms.10 Our case that had CD with abdominal protrusion, vomiting and poor feeding, manifested just after birth with abdominal protrusion. It is the first reported case of CD in Iran as manifested in the neonatal period. Therefore, although CD is rare, it should be considered in the differential diagnosis of abdominal masses, abdominal protrusion, icter and congenital anomalies of the heart and kidneys. The authors thank A. Tabatabayee, MD, for his review of our pathologic specimen.
Copyright 2004 -Medical Journal of the Islamic Republic of Iran The following images related to this document are available:Photo images[mr04034f1.jpg] [mr04034t1.jpg] [mr04034f3.jpg] [mr04034f2.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}