 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
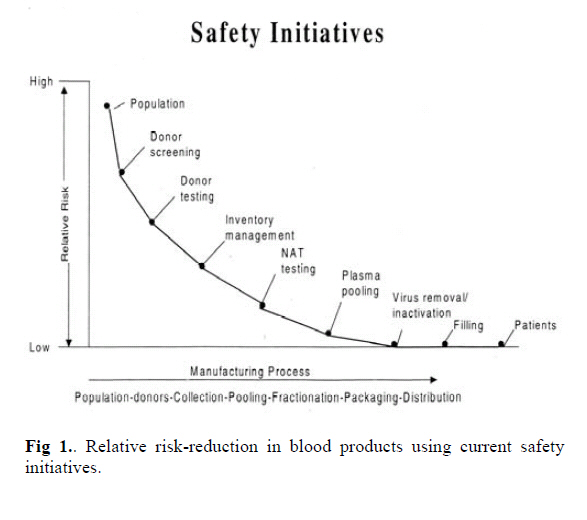
Medical Journal of the Islamic Republic of Iran , Vol. 20, No. 2, July, 2006, pp. 86-92 SAFETY OF BLOOD AND PLASMA DERIVATIVES: PATHOGEN REDUCING TECHNOLOGIES H. REZVAN, Z. MOTALLEBI, M. A. JALILI, K. MOUSAVI HOSSEINI AND A. A. POURFATHOLLAH* From the Iranian Blood Transfusion Organisation -Research Center, IBRF R&D Dept., and the *Immunology Dept. of Tarbiat Modarress University, Tehran, Iran. Code Number: mr06019 ABSTRACT Human blood and blood products is the source of a wide range of medicinal products used for the treatment and prevention of a variety of injuries and disease. Despite stringent routine measures and filters employed, residual pathogen infectivity remains an important challenge in the field of blood transfusion. In this article various measures and technologies that can be applied in order to reduce the residual risk are reviewed. Keywords: Plasma derivatives ;Virus inactivation; blood borne viruses. INTRODUCTION Human blood is the source of a wide variety of medicinal products used for treatment and prevention of various diseases. Over the past two decades, blood transfusion in general and plasma fractionation in particular have developed into medical and scientific disciplines requiring skilled scientific expertise and advanced technology to prevent transmission of blood -borne infections. Appropriate measures to prevent and reduce the risk of potential infections have been introduced with implementation of strict regulations for every step of production from donor selection right up to follow-up of the clinical use of end products. In general, blood products are of two major categories: (a) the single donor products such as red cell concentrate, platelet concentrate, leucocytes and single donor fresh frozen plasma (FFP). (b) Plasma derivatives, which in contrast to single donor components are obtained from pooling thousands of plasma donations and require a manufacturing approach that includes specific measures to ensure the very high margin of safety demanded by society and regulatory bodies. Fig. 1 demonstrates the relative reduction of risk factor by taking various "safety initiatives". These plasma derivatives are comprised of an increasing variety of products including albumin, various coagulation factor concentrates, polyvalent or hyperimmune immunoglobulins, protease inhibitors, anticoagulants and fibrin sealants. Viral safety of blood products relies mainly on the appropriate selection of donor population and on the quality of the viral screening procedures for each donation. However stringent, these systems are not capable of excluding a unit of blood or plasma being collected from a donor in an infectious window period when routine screening tests are negative but viral infectious particles may be present at very low concentrations. This residual infectious risk which in developed countries is very low, becomes more significant when subsequent pooling is used to manufacture plasma products, because all resulting products from an infected pool may be infectious to varying degrees and potentially may infect a large population of recipients. The diversity in the type of blood and plasma products illustrates the need for a "customized" strategy to ensure optimum viral safety for each product. It also underlines a complex sustaining regulatory process that governs blood and plasma derivative production worldwide. Fully mastering the relative risks of such diverse products remains a major challenge in this field. This paper is aimed to provide an update and insight to the wide range of measures designed to enhance the safety of plasma products and to provide relative data and experience available in our country where applicable. Infectious Risks Human blood can be contaminated by various infectious agents, viruses, bacteria, protozoa and prions. The major viruses of concern are Hepatitis B virus (HBV), Hepatitis C virus (HCV) and Human Immunodeficiency virus (HIV). Other important viruses are the Human T-cell leukemia virus (HTLV-I,II), Parvovirus B19, Herpes viruses including CMV and Hepatitis A virus (HAV).1 More recently, a list of additional viruses such as GBV-c, West Nile Virus (WNV), TTV, etc…have emerged as potential threats.2 Despite continuous improvement of screening tests, transmission of viral infections by blood and products remains a major challenge and the risk is not totally excluded, since the current tests are still unable to detect infection in the window period today, in countries where donor selection and donor screening is implemented the residual risks are very low. Frequencies of 1 in 2.3 million for HIV and 1:620,000 and 1 in 400,000 for HCV and HBV has been reported for Europe.3 Accordingly estimates of the frequency in France and in Germany is reported separately as 1 : 1 million for HIV, 1: 200,000 for HCV and 1:180,000for HBV.4 Similar although slightly higher figures were reported in the U.S.A.5,6 Evidently, in countries with a higher prevalence of blood borne viruses, and less rigorous donor exclusion, the risk rises and will be higher. In Iran with prevalence rates of HCV at around 0.3%, 7 and HIV being close to the European countries and use of third generation Elisa techniques for screening, the residual risk factor for these infections should be close to that reported in Europe. However, in the case of HBV, the prevalence rate amongst blood donors in Iran is much higher than the European countries, an average 3% HBsAg positivity among the blood donors reported in 1994 is reduced to 1.2% in 20038 by employing more rigorous donor selection programmes, therefore the residual risk in Iran should be higher than that reported for the European countries. Another important threat is bacterial contamination of cellular products. In some countries, bacterial sepsis today appears to be the most frequent infectious complication, especially of platelet transfusions, but the frequency of transfusion associated septic reactions varies widely in Europe up to six cases are reported annually in European countries and in Canada. In the USA one septic reaction is observed per 10000-20000 transfused units of platelet concentrate (PC) and fatal reactions are seen in one per 6 million units. 9 Bacterial contamination of red blood cell concentrates (RBC) also occurs frequently.10 In the period of 1986 to 1996 a total of 21 cases of sepsis associated with contaminated RBC were reported in the USA.11 Various parasitic diseases may also be transmitted by blood products, of greatest concern are post transfusion malaria and particularly Chagas disease.1 Finally, prions that is the agent of Creutzfeldt Jakob Disease (CJD) and its new variant (vCJD) is also a potential hazard which calls for concern. Citing two confirmed cases in the United Kingdom where v-CJD was transmitted from blood donors who were young and apparently healthy, is a warning for a new wave of infections of unknown magnitude around the world.12 As illustrated risk of transmission of bacterial and parasitic pathogens is limited to the single unit products, however in the case of plasma products due to having filtration steps using 2 µm filters these larger pathogens are removed but viruses and prions are of great concern. The risk of contamination derives from the balance between the viral load in the starting plasma pool, the presence of any neutralizing antibodies, the ability of the manufacturing process to reduce the risks, and the patients own physiological condition. In that respect, an extensive knowledge of the epidemiology of blood borne viruses in every society is of vital importance. For example although transmission of HAV through blood and its products is important in the Western societies where rate of exposure to this virus is not high in other countries such as Iran where 90% of adults have been exposed to the virus and carry neutralising antibodies to HAV13, the high titre of antibody present in plasma products with local origin will neutralize any small residual risk of contamination with this virus, therefore lowering concern. In other words according to the endemicity of any infection specific safety strategies should be considered by the regulatory bodies in various countries. In the past 20 years, more sophisticated production processes and advanced virus inactivation procedures have provided the means to reduce and prevent infections related to HBV, HCV, and HIV. More recently the progressive introduction of nucleic and amplification testing (NAT) for detection of viral genomes which closes the window period considerably provides a new dimension in the viral safety of blood and plasma derivatives.14 Use of viral inactivation procedures in blood safety Specific viral inactivation treatments are the corner- stone in ensuring a sufficient margin of safety of plasma products. The major target of the antiviral treatments is the inactivation of the highly pathogenic plasma -borne enveloped viruses that is, HIV, HBV and HCV which cause chronic infection and disease. Additional targets are the less pathogenic non-enveloped viruses such as HAV and B19, which cause mainly acute illnesses. The various procedures applicable to plasma products is briefly described below. However, because of their cytotoxic properties, these methods cannot be applied to cellular products. The limited available procedures are discussed in a separate section. I- Viral inactivation procedures of plasma derivativesPasteurization Pasteurization is defined as the treatment of a liquid protein for 10h at 60ºC, the mechanism of viral inactivation using this procedure is heat denaturation of viral components, thereby inhibiting virus replication. This procedure is generally performed in the presence of stabilizers, which are added to protect plasma protein biological function and limit molecular alterations. The stabilizers may have a protective effect that is specific to plasma proteins, this is the case for albumin where sodium caprylate and sodium tryptophanate bind to the protein and prevent its denaturation or aggregation during liquid heat treatment.15,16 Other stabilizers like sugars (e,g. sucrose), and polyols (e.g. sorbitol), have a more global protective effect on the biological activity of labile proteins, avoid the formation of protein aggregates and limit risk of precipitation.17, 18 Amino acids (e.g. lysine, glycine, arginine, etc…) may also protect the functional activity of some plasma proteins.18, 19, 20 Combination of sugars and amino acids has been successfully used for the pasteurization of Factor VIII 16 ,19,20, 21, Factor VII.22 Other plasma products that can be pasteurized are intravenous IgG23, IgM-enriched IgG24, where possibility of pasteurization in absence of stabilizers is reported, also anti-thrombin III and alpha-1 antitrypsin.25,26 Stabilisers when added prior to heat treatment can be subsequently removed by precipitation ultrafiltration, chromatography or a combination of those. Manufacturing processes must ensure consistent and homogenous application of the heating process, and avoid risk of subsequent contamination. Presently albumin is the only product which can withstand pasteurization in the final container, since the quantity of stabilizers used is compatible with direct clinical use and therefore alleviates the risk of downstream contamination. Extensive validation studies has shown that pasteurization is capable of inactivating various viruses, whether enveloped or non-enveloped, including HIV, HBV, HCV and HAV.21 ,26 The confidence in the efficacy of pasteurization for virus inactivation is linked to the remarkable record of safety for albumin.27 More recently, inactivation of WNV using this procedure has also been reported.28 Dry heatDuring this treatment a lyophilized product in the final container is subject to heating for several minutes to several days at temperatures from 60 to 100ºC.21, 26, 27 Initially, dry heat treatment at 60 to 68ºC for 72 to 96 h was applied to coagulation factors with objective of inactivating HIV. However, reliable inactivation of HCV in these concentrates is achieved only by more severe conditions at 80ºC for 3 days.29 There are other reports where heat treatment at 100ºC for 30 min has been applied to Factor VIII concentrates in order to inactivate HAV, although, the transmission of B19 may not be eliminated.30, 31 The viral inactivation efficacy of heat is generally less than pasteurization due to lower moisture content of the product. Steam heatAt equivalent temperatures, a higher level of virus inactivation can be achieved by the addition of steam and pressure, and a relatively high moisture content. The extent of viral inactivation is directly related to the temperature (60 to 80ºC), the pressure (1190 to 1375 mbar) and the duration of treatment (upto10h.). Vapor heating was shown to inactivate HAV in plasma products32, but cases of transmission of HCV through coagulation factor has been reported33,34, although the preponderance of clinical data indicates safety with respect to hepatitis viruses and HIV.35 Solvent / detergent treatmentOrganic solvent / detergent mixtures disrupt the lipid membrane of enveloped viruses. Once disrupted the virus no longer can bind to and infect host cells. Non-enveloped viruses are, therefore, not inactivated using this procedure. The organic solvent, tri (n-butyl) phosphate (TNBP) is used at a concentration of 0.3% and 1% non ionic detergent, either Tween 80 or Triton x-100 at 24ºC for a minimum of 4h with Triton and 6h for Tween.36, 37 The elimination of the viral inactivation agents is achieved by subsequent ultrafiltration, chromatography or precipitation techniques. Permitted residual levels of TNBP, Tween 80 and Triton x-100 typically are 3-25, 10-100 and 3-25 ppm, respectively.21 The method has proven to be extremely robust and manufacturing processes have minimal effect on its efficiency.38 This method has been applied to coagulation factors VIII39, 40, IX41, 42, factor VII43, immunoglobulins44 although in the case of immunoglobulins a new alternative using caprylate has also been introduced.45 S/D treatment has been shown to have unchallenged potential to inactivate enveloped viruses while maintaining plasma protein integrity and represented a breakthrough in the viral safety of plasma products. Enveloped viruses are typically killed in less than a few minutes. There is no report of protein neoantigen formation due to SD treatment and there is excellent protein recovery. The method used to remove the SD agents may induce some protein loss. A drawback of the SD procedure is the failure to inactivate non-enveloped viruses. To date, there has been no report of transmission of HIV, HCV or HBV by SD-treated plasma products in about twenty years of use. The efficacy of this procedure in inactivation of an emergent enveloped virus such as WNV has been demonstrated.28 Cases of transmission of non-enveloped viruses such as HAV and B19 have been reported from SD-treated coagulation factor concentrates.46, 47 Acid pHSome of the purification steps involved in the production of plasma derivatives have proved to act as viral inactivation steps. The case of pH 4 treatment of IgG which was developed initially to reduce anti- complementary activity and aggregation is a good example. Incubation of IgG at pH 4 and at temperatures between 30 to 37ºC for more than 20 hours inactivates substantial dose of enveloped virus.48,49 Complete inactivation (over 5 to 8 log) of HIV and other enveloped viruses is achieved by contrast; non-enveloped viruses are more resistant.21 Specific viral elimination treatment: NanofiltrationBy contrast to fractionation technologies that remove viruses in a non-specific mechanism, nanofiltration is a new approach to eliminate microorganisms specifically. Nanofiltration is carried out on membranes of a few nanometers (15 to 45 nm) and works by the segregation of proteins and viruses on the basis of their size.50, 51 Therefore, biological infectious agents such as viruses larger than the nominal filter porosity are retained on the membrane while smaller plasma proteins pass through. Compared with other viral reduction means, nanofiltration may be the only method permitting efficient removal of enveloped as well as non-enveloped viruses, under conditions where 90-95% of protein activity is recovered. 52 ,53 The technique is performed using commercially available cartridges and is reasonably simple. In fact, similar to aseptic filtration for removal of bacteria it can be implemented without the need for sophisticated equipment. Viral validation studies have revealed that membranes with porosity in the range of 15 to 40 nm. can be used for all plasma products. As expected viral clearance of small non-enveloped viruses is greater with the 15 nmcartridge, although aggregates of these viruses have successfully been removed using 35 nm. Filters.53 Preliminary data indicate that nanofiltration may also remove prions and is therefore becoming increasingly a routine step in manufacture of biopharmaceuticals.54 ,55 Application of nanofiltration has been reported for production of factor IX concentrates41 ,56 ,57,58 ,intravenous immunoglobulins59,60, factor VIII concentrates61, and activated factor VII concentrate.62 II-Viral inactivation of single donor productsMost of viral inactivation procedures to date are limited to red cell concentrates, platelet concentrates and fresh frozen plasma (FFP). Since the addition of any compounds to these products may affect their functional properties, an assessment of their quality before and after pathogen inactivation is of crucial importance. In addition, their safety and efficacy have to be studied in vivo in animals and humans. Methods that are successfully applied in clinical medicine are as follows: Solvent / detergent (S/D) treatment of FFPAs mentioned in the previous section, the lipid envelop of transfusion relevant viruses is destroyed by a combination of an organic solvent and a non-ionized detergent, whereas non-enveloped viruses are not affected. Few studies showed that structure and biological function of most plasma proteins were well preserved.63, 64 However, these reports show that some sensitive proteins such as factor VIII, Protein S and alpha-2- antitrypsin may be slightly damaged. For the production of S/D treated FFP, the organic solvent TNBP is combined with Triton-X-100, at 30 Computer for 4h and the S/D reagents are then removed by extraction. The plasma is sterile-filtered and filled into plastic bags or glass vials in 200 mL units and at -30 C or lower. Since 1991, S/D treated FFP has been clinically used in some countries and has good efficacy and safety record.65 However, contamination by non-enveloped viruses may pose a problem. The S/D technology cannot be applied to cellular blood products, since the reagents would disintegrate the lipid bilayer of the cell membrane and destroy the cells. Methylene blue treatment of FFPThe virus inactivating properties of Methylene Blue (MB) in combination with visible light have been known for a long time. Researchers at the German Red Cross Blood Transfusion Service have developed a procedure using this property of MB for inactivation of FFP. In this procedure 1 µmoL. of MB (320 ug/L) is added to single donor units of FFP, and illuminated with visible light.66 This treatment inactivates enveloped viruses, HBV, HCV and HIV. Since 1992, this blood product has been used on large scale in Europe.67 The Paul Ehrlich Institute has expressed concern about possible mutagenic properties of MB and removed approval in Germany. Leucocyte depletionA widely used approach for removing leucocytes is the" buffy coat" procedure, where the bulk of leucocytes (buffy coat) is segregated from the plasma and the cells and transferred into a special buffy coat bag. This method allows removal of 85% of the leucocytes. The other method in common use is employing leucocyte filters. Development of several generations of leucocytes filters made it possible to reduce leucocytes to very low levels; leucocyte filters currently used are capable of reducing 5log of white cells in a one step procedure with a minimal loss of red cells. Leucocyte depletion of platelet concentrates obtained by platelet apheresis from single donors may be achieved by centrifugation methods or by leucocyte reduction filter systems. Leucocyte filtration may be performed at the bedside, that is, immediately before the transfusion. This method allows specific targeting of leukodepleted products to patients who need them. In recent years, animal experiments have suggested that leucocytes may be involved in promoting v-CJD68, thus in order to minimize a potential risk, some European countries have introduced universal leukodepletion of all labile products.67 Although appropriate prestorage leucocyte depletion by filtration was shown to prevent some cell-bound infectious agents such as CMV, studies demonstrating that this technology also excludes cell-bound HIV and HTLV-I,II infectivity are still lacking. The high level of safety achieved using the very complicated and expensive technologies for plasma products should be an encouragement to seek further progress and indeed, new treatments continue to be developed. However, considering the high relative safety of blood in the industrialized world, simple, less expensive viral safety treatments of blood and plasma derivatives in the developing world appears to be urgent and challenging. To this date the only available methods of viral inactivation employed in the production of plasma derivatives in Iran has been pasteurization and mild heat treatment. The other more sophisticated methods such as severe heat treatment and solvent /detergent have been applied to such products only in laboratory scale. The technology for the proper application of these techniques to such products in industrial scale is not as yet available. REFERENCES
Copyright 2006 -Medical Journal of the Islamic Republic of Iran The following images related to this document are available:Photo images[mr06020f1.jpg] |
| |||||||||
{kind=link}