 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Medical Science Vol. 58 No. 2, February 2004 , pp. 67-71 Case Report Arthritis in Lymphomatoid granulomatosis: Report of a case and review of literature Vikas Agarwal, Amita Aggarwal,* Lily Pal,** Ramnath Misra* DM Senior Lecturer; DM;* MD;** MD;* Department of Medicine, Government Medical College & Hospital, Sector 32, Chandigarh, India; Department of *Clinical Immunology & **Pathology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, India
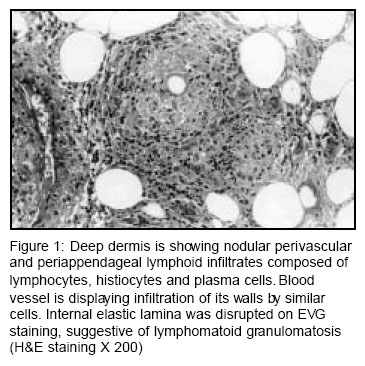
Accepted Date: 12-02-2004 Code Number: ms04010 Abstract Lymphomatoid granulomatosis (LG) is a rare systemic vasculitis caused by Epstein Barr virus induced transformation of the B-cells in a T-cell rich environment. The predominant clinical presentations are confined to the pulmonary system however; extra-pulmonary manifestations can sometimes be the main feature of the disease. Here in we describe a 52-year-old female who presented with symmetric polyarthritis and generalized stiffness for 7 months and papular lesions over extremities for 3 months duration. She in addition had generalized lymphadenopathy. Histopathological examination of the cutaneous lesions confirmed LG. Patient died despite therapy with cyclophosphamide and prednisolone. This is the first report of LG mimicking rheumatoid arthritis from India. Key Words: Rheumatoid, Angiocentric lymphoma, Epstein Barr virus. Introduction Lymphomatoid granulomatosis (LG) is a rare multisystem disorder characterized by a focal and transmural angiocentric and angiodestructive pleomorphic cellular infiltration.1 Historically, it was believed to be a post thymic T-cell disorder.2 Recently, Epstein Barr virus induced transformation of the B-cells has been proven to be the main pathology.3 Arthritis in LG has been described rarely. Herein we are discussing a case of LG presenting with polyarthritis mimicking rheumatoid arthritis (RA). Case Report A 52-year-old female presented with bilateral symmetrical inflammatory polyarthritis involving Shoulder, elbow, wrist, metacarpo-phalangeal (MCP), proximal interphalangeal (PIP), knee, ankle, and metatarso-phalangeal joints and generalized stiffness of shoulder and pelvic girdle muscles for last 7 months and recurrent crops of erythematous papular skin rash involving all four limbs of 3 months duration. Stiffness and polyarthritis used to respond dramatically to low dose prednisolone (10 mg/day). There was no history of temporal or occipital headaches, jaw claudication or visual disturbances. She had diabetes mellitus for the last 7 years. Examination showed erythematous maculo-papular rash over all 4 limbs, synovitis of the above mentioned joints, moderate hypertension and bilateral cervical and axillary lymphadenopathy. Rest of the examination was unremarkable. Investigations revealed hemoglobin 10.6 gm/dl, erythrocyte sedimentation rate (ESR) 36 mm, C-reactive protein (CRP) 5.65 mg/dl (normal <0.5), polyclonal hypergammaglobulinemia and fasting blood sugar 202 mg/dl. Urinalysis showed proteinuria (24 hour urine protein 640 mg) without active sediments. Tests for antinuclear antibody, antineutrophilic cytoplasmic antibody, rheumatoid factor, renal and liver function tests were within normal limits. X-ray of the hands was normal however, X-ray chest revealed right paratracheal lymphadenopathy. Computerized tomographic scan of the thorax showed mediastinal and hilar lymphadenopathy. Fine needle aspiration cytology (FNAC) from the right axillary and mediastinal lymph node showed reactive hyperplasia. Skin biopsy from the right lower limb papular lesions revealed normal epidermis and superficial dermis. Deep dermis showed nodular perivascular and periappendageal polymorphic infiltrates composed of lymphocytes, histiocytes and plasma cells. At places this polymorphic infiltrate was spreading to the subcutis. Many blood vessels in the deep dermis and the subcutis were infiltrated by similar cells with disruption of the internal elastic lamina (EVG staining), suggestive of LG (Figure 1). Immunohistochemical analysis of the tissue revealed predominance of CD4+ T cells without any reactivity with the B-cell markers. She was treated with cyclophosphamide and prednisolone (1mg/kg/day). Three months later she presented in hypotension with dyspnea, tachypnea and altered sensorium. Her hemogram, renal and liver functions were normal and cultures (blood, urine) were negative for infection. Skiagram chest revealed bilateral basal reticulonodular shadows. She died after 36 hours of hospitalization. Discussion The diagnosis of LG requires demonstration of polymorphic angiocentric and angioinvasive infiltrate (1) as was seen in our case. Lung is the most common organ to be involved in LG and it characteristically manifests in form of nodules, cavities, infiltrates, consolidation and pleural thickening.1,4 Mediastinal lymphadenopathy is uncommon and is more in favour of evolution to lymphoma,4,5 however, FNAC from the axillary and mediastinal lymph node in our case showed reactive hyperplasia only. Skin involvement, the second most common affected organ in LG,6 has been reported in up to 55% cases.5 Scattered nodules are the most commonly observed lesions however, other cutaneous manifestations like papules ulcers, plaques, eroded and crusted lesions, facial oedema, and folliculitis-like eruptions, necrobiosis lipoidica, lobular panniculitis, maculo-papular rash macular erythema and annular infiltrated lesions with central clearing mimicking Hansen's disease have been reported.6 The majority of the cutaneous lesions mirrors to some extent LG in the lung, although EBV+ cells are less frequently identified.6 Our inability to recognize B-cells in the angiocentric infiltrate in skin biopsy tissue may be due to the scarcity of the B-cells in these lesions or the overwhelming presence of the T-cells as has been reported earlier.3 The differential diagnoses of these lesions like sarcoidosis, lymphomatoid papulosis, Wegener's granulomatosis and lymphoma can be distinguished by the characteristic histopathology of the respective lesions. Skin biopsy in our case revealed angioinvasion and angiodestruction alongwith polymorphic infiltrate involving appendages in the dermis and subcutis, which strongly favored the diagnosis of LG. James and colleagues,7 reviewed 22 skin biopsy slides, of 44 cases of LG with cutaneous manifestations, and reported that angiocentric and angioinvasive vasculitis with polymorphic infiltrate was present in small number of cases, polymorphonuclear leukocytes and multinucleated giant cells were rare and mixture of mature lymphocytes, plasmacytoid cells, plasma cells, histiocytes and atypical lymphoreticular cells were common. They noted frequent subcutaneous tissue involvement. Involvement of dermal appendages and nerves was seen in high percentage of cases. Arthralgia has been reported in 6-20% of patients with LG,1,4 however, arthritis is unusual. In the series by Leibow, of 40 patients only one had arthritis.1 Arthritis in this patient preceded the diagnosis of LG by 3 years and therefore seemed unrelated. In the largest series till date comprising of 152 patients, one patient with rheumatoid arthritis (RA) was reported to develop LG.4 Otherwise there are only a few case reports of arthritis in LG.8-10 The clinical presentations of all the cases have been summarized in Table 1. Arthritis may be a presenting feature,8,10 or may antedate the systemic manifestations by almost 3 years.1,9 In an earlier report, a 26 years young male was described to present with fever, rash, lung consolidation and polyarthritis. The joint distribution mimicked RA. Arthritis responded poorly to non-steroidal anti-inflammatory drugs (NSAIDs) but had good response to prednisolone.8 Whereas in two other reports polyarthritis mimicked RA and had good response to NSAIDs.9,10 In none of the three reports joint erosions and rheumatoid factor were detected. The pattern of joint involvement in our case too mimicked RA and was responsive to steroids as has been reported earlier.8-10 While the three patients reported with arthritis in LG till date were males ours is the first female patient of LG presenting with polyarthritis. Though the exact pathogenesis of arthritis in LG is not clear Bergin et al, have demonstrated lack of immune complexes and complement activation in the serum, predominance of lymphocytes in the synovial fluid, and perivascular lymphoid infiltrate with extension into walls of the small vessels in the synovial histology from the knee joint.8 It might be argued that injury of the synovial vessels by lymphohistiocytic infiltrate may be responsible for polyarthritis in LG. Our case highlights the need to consider the differential diagnosis of LG in patients presenting with triad of polyarthritis, lung and cutaneous manifestations. References
Copyright by The Indian Journal of Medical Sciences The following images related to this document are available:Photo images[ms04010f1.jpg] [ms04010t1.jpg] |
| |||||||||

{kind=link}
{kind=link}