 |
search
for |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Medical Science Vol. 58 No. 5, May, 2004 , pp. 185-190 A COMPARATIVE STUDY ON IDIOPATHIC PULMONARY FIBROSIS AND SECONDARY DIFFUSE PARENCHYMAL LUNG DISEASE H S SUBHASH, I ASHWIN, S K SOLOMON,*,** T DAVID, A M CHERIAN,***,**** K THOMAS*** MD; **MBBS; ****FRACP. *Registrar; Lecturer; ***Professor; Department
of Medicine Unit 2,
Christian Medical College and Hospital, Vellore, India.
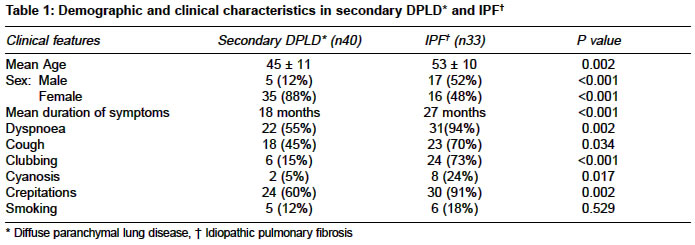
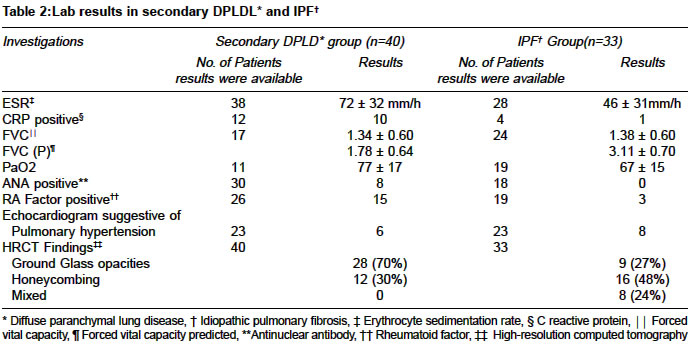
Code Number: ms04033 ABSTRACT BACKGROUND AND AIMS: Clinical characteristics of patients diagnosed to have Diffuse parenchymal lung disease (DPLD) were evaluated in this study. DESIGN AND SETTING: Retrospective evaluation, a tertiary care center in South India. Material and METHOD: Subjects diagnosed to have DPLD over a five-year period were included in this study. Data pertained to clinical characteristics and lab parameters were obtained. STATISTICAL CONSIDERATIONS: t- test for Mean values and chi-square test for comparing proportions were used. RESULTS: There were 73 eligible patients included for evaluation. Secondary cause for DPLD was diagnosed in 40 (55%) and idiopathic pulmonary fibrosis (IPF) was diagnosed in 33 (45%). The mean age was 45±11 and 53±10 years, of these 5 (12%) and 17 (52%) were male subjects in the secondary DPLD and IPF group respectively. The mean age, dyspnoea, cough, clubbing and crepitations were noted to be higher in patients with IPF as compared to patients with secondary DPLD. Fifty patients were followed up for a mean of 13 months (28 secondary DPLD and 18 IPF). Follow up data was available in 46 patients. Of these subjects prednisone alone was initiated in 24 subjects and combination with azathioprine in 22. Subjective improvement in symptoms was noted in 29/46 (63%), 19 with secondary DPLD and 10 with IPF. CONCLUSION: symptoms and signs were noted more frequently with IPF, subjective improvement to treatment was noted in 63% and the best response was noted among patients diagnosed to have sarcoidosis. A prospective trial is needed to study the long term prognosis and therapeutic response among Indian patients. KEY WORDS: Idiopathic pulmonary fibrosis, Cryptogenic pulmonary fibrosis, Collagen vascular disease, Prednisone, Azathioprine. INTRODUCTION Diffuse parenchymal lung disease (DPLD) is one of the important disabling disorders of the lung and it is being increasingly recognised in India.1-7 There are few published reports on the various etiological factors responsible for DPLD from Indian series.1-6 However there are number of published data in Indian literature on various other aspects of DPLD. Though a definitive diagnosis of DPLD requires lung biopsy, it is being diagnosed with a fair degree of confidence on the basis of clinical and radiological criteria alone without histological confirmation.1,8 Various classification of DPLD is described in recent literature.9 Most published data available in the literature on DPLD are from the west. There is paucity of data among Indian subjects with DPLD regarding long-term prognosis, survival and therapeutic response to steroids or immunosupression drugs. In this study we evaluated the clinical and laboratory features of patients diagnosed to have DPLD. We have also made an attempt to study the clinical course of these patients for whom follow up data was available. MATERIAL AND METHODS This retrospective study was conducted in Christian Medical College and Hospital, a tertiary care 1800-bed university teaching hospital in south India. All subjects diagnosed to have DPLD in one General medical unit over a five-year period were included in the study. Patients diagnosed to have DPLD were identified by review of admission/discharge and out patient records. Diagnosis of DPLD was made if clinical / pulmonary function tests were suggestive and High Resolution CT Scan (HRCT) was consistent of DPLD in all subjects. The case records were retrieved from the medical records department and data was extracted to obtain information on age, gender, duration of symptoms related to the lung disease, clinical signs and symptoms as recorded by the treating physician. Data was also obtained on results of blood gas analysis on breathing room air, pulmonary function test results and other tests done towards the diagnosis of etiology of secondary causes of DPLD. The findings of HRCT were assessed for the presence of ground glass opacities or honey combing of the lung. Results of echocardiogram findings were assessed for the presence or absence of pulmonary hypertension secondary to DPLD. Patients were considered to have a Secondary cause of DPLD if they were diagnosed to have collagen vascular disease (confirmed by serologic and other standard criteria), other systemic diseases, occupational or environment exposure known to cause interstitial pulmonary fibrosis. Patients without any identifiable cause for DPLD were considered to have idiopathic pulmonary fibrosis (IPF). The duration of follow up was defined as the interval between diagnosis of DPLD and the last known contact in our center. During the follow-up visit presence or absence of symptoms related to DPLD as judged by the treating physician and laboratory parameters were recorded when available. STATISTICAL CONSIDERATIONS Mean values were compared using t- test and chi-square test was used for comparing proportions. RESULTS Demographic, clinical and lab results There were total of 97 patients with the diagnosis of DPLD during the five-year period, of them 24 subjects were excluded from the study either because HRCT results were unavailable for analysis or other concomitant diagnosis was considered during the follow up period. Secondary cause of DPLD was diagnosed in 40 (55%) subjects. Idiopathic pulmonary fibrosis was diagnosed in 33 (45%) patients. Comparison of the demographic and clinical characteristics among patients with secondary DPLD and IPF are given in Table 1. Among those subjects with secondary cause of DPLD systemic sclerosis was diagnosed in 12, rheumatoid arthritis in 11, overlap collagen vascular disease in 8, sarcoidosis in 7 (diagnosed by tissue biopsy) and Systemic lupus erythematosis in 2. The comparison of the lab results among patients with secondary DPLD and IPF are given in Table 2. Treatment and follow up results Fifty Patients were followed up for a mean duration of 13 months (range 2 to > 60 months) and follow up clinical data was available in 46 patients, 28 with secondary DPLD and 18 with IPF. Among those patients who were followed up prednisone was initiated in 24 subjects, combination of prednisone and azathioprine was initiated in 22 subjects. In addition to the above drugs chloroquine was given in 11, salazopyrine in 11, methotrexate in 6, cyclophosphamide in 2, D. Penicillamine in 2, and cyclosporine in 1 for their underlying collagen vascular diseases. Subjective improvement in symptoms was noted in 29/46 (63%), 19 with secondary DPLD and 10 with IPF. The best response to treatment was noted in patients with sarcoidosis 5/7 (70%). The symptoms remained same or worsened in 17/46 (37%) subjects, 9 with secondary DPLD and 8 with IPF. To assess the objective improvement of the lung functions follow-up spirometry results were available in only 36 % of the subjects. The follow up FVC results was 1.32 ± 0.42 L among patients with secondary DPLD and 1.39 ± 0.68 L among patients with IPF. DISCUSSION In the present study IPF was noted in 45% and DPLD secondary to underlying diseases was noted in 55%, which has remained almost similar to the earlier published reports from India.2,3 The mean age of patients diagnosed to have IPF was more than 50 years in this present study contrary to an earlier report from north India2 were the mean age was noted to be around 40 years among patients with IPF. Clinical features such as dyspnoea, cough, clubbing and crepitations were noted more frequently among patients with IPF compared to those with secondary DPLD in this present study. Ground glass attenuation on HRCT was predominantly noted among patients with secondary DPLD in this present study. Ausculatory evidence of pulmonary hypertension among patients with interstitial pulmonary fibrosis is reported to occur in about 70%.8 Jindal3 et al reported pulmonary hypertension in 38% among patients with DPLD on the basis of clinical, radiological and electrocardiogram findings. In the present study echocardiographic evidence of pulmonary hypertension was noted in 14/46 (30%) of the patients. Standard lung function tests are recommended in assessing disease activity among patients with IPF.11 The mean vital capacity among Indian subjects with DPLD is noted to be less than 55 to 61% for the predicted value.2,3 In the present study of the available data there was no difference noted in the mean vital capacity among patients with IPF and secondary DPLD. Most forms of DPLD rarely remit and feature periods of exacerbation superimposed on chronic or worsening baseline symptoms.12 Most earlier published reports on DPLD has shown to have a overall 40 to 70% subjective improvement to treatment with corticosteroids and immunosuppressive agents such as azathioprine, cyclophosphamide and others but objective improvement is noted to occur in only 12 to 30%.8 Behera et al6 in their retrospective study among patients with IPF reported 48 % subjective improvement treated with prednisone. More resent systematic review among patients with IPF has shown no convincing evidence to suggest corticosteroids are helpful in the management of IPF.13 However there is some evidence to suggest that there may be long-term survival advantage with azathioprine in the management of patients with IPF.14-16 Usefulness of azathioprine in the management among Indian patients with DPLD is not been well documented earlier. In the present study subjective improvement was noted in 63% of patients treated with steroids and azathioprine. The best response to treatment was noted among subjects diagnosed to have sarcoidosis. Al Mobeireek17 has reported similar findings among patients DPLD secondary to sarcoidosis. In a study with a two-year follow up, lung function in patients with IPF was noted to be deteriorated significantly compared to those with DPLD secondary to collagen vascular disease.18 In this present study we could not assess the lung functions in all the subjects who were followed up because of insufficient data. However of the available data in 36% there was no much change in the mean follow up FVC compared to the initial FVC. More importantly there was no deterioration noted in the follow up FVC with treatment. The limitation of this study was that test results were not available in all patients and only subjective response to treatment could be assessed during the follow up visits because of unavailability of sufficient lab data to assess the lung functions objectively. Understandably this is an inherent problem of a retrospective study design. Most published data available in the literature on DPLD are from the west. Since there are different environment and genetic factors in Indian sub continent compared to the west prospective trial will be needed to document the therapeutic response with immunosupression and immunomodulatory drugs, exact long term prognosis and survival among Indian patients with DPLD. REFERENCES
Copyright by The Indian Journal of Medical Sciences The following images related to this document are available:Photo images[ms04033t2.jpg] [ms04033t1.jpg] |
| |||||||||

{kind=link}
{kind=link}