 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Medical Sciences, Vol. 59, No. 9, September, 2005, pp. 407-417 Practitioners section Genetics of mental retardation Ahuja AS, Thapar Anita, Owen MJ Department of Psychological Medicine, Cardiff University,
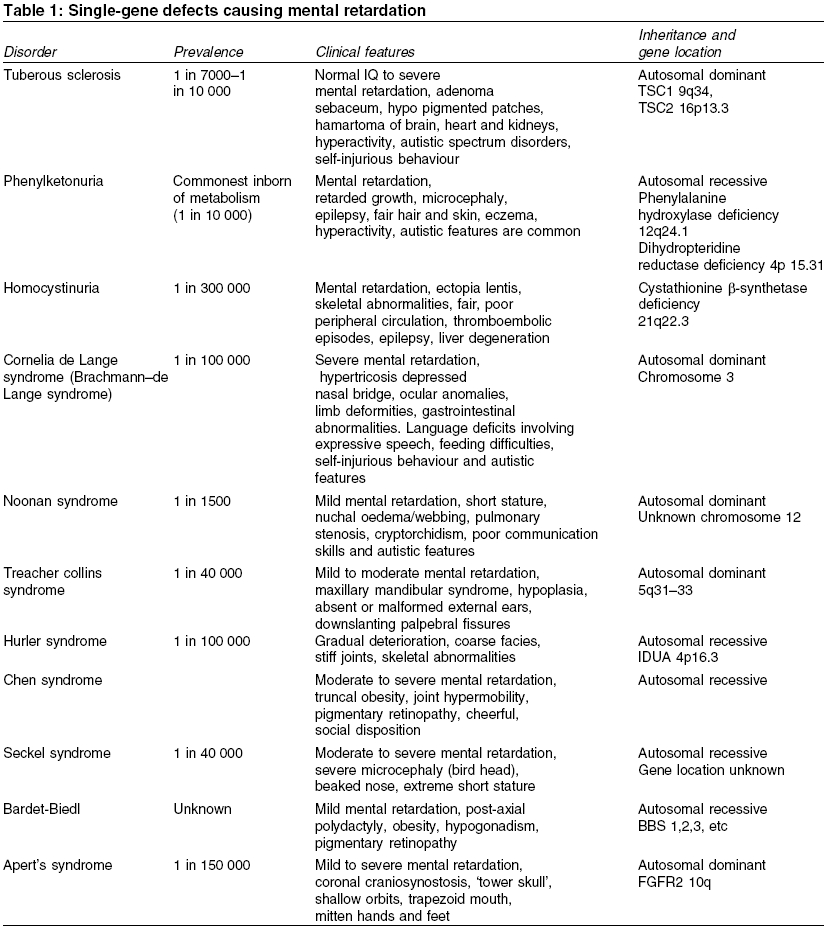
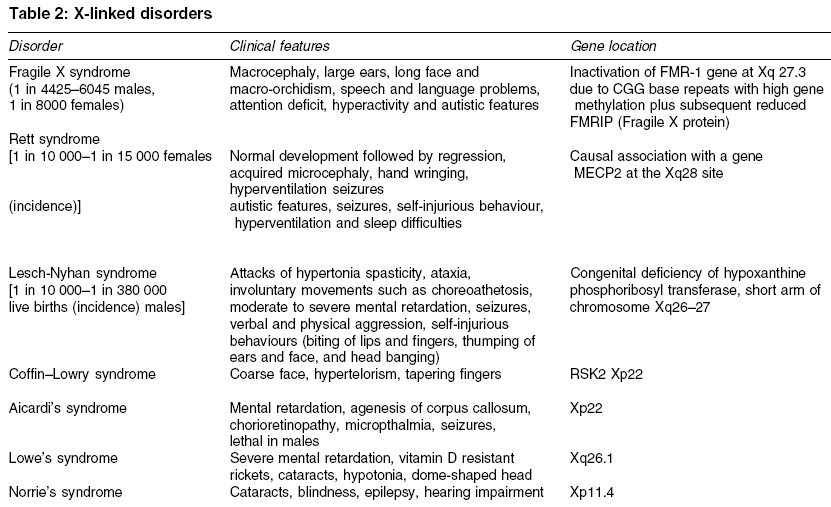
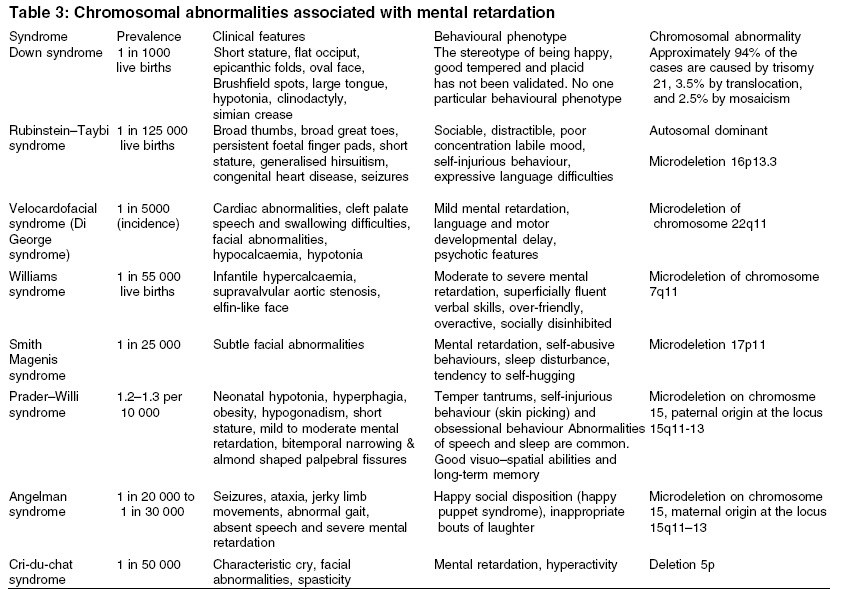
Heath Park, Cardiff Code Number: ms05063 ABSTRACT Mental retardation can follow any of the biological, environmental and psychological events that are capable of producing deficits in cognitive functions. Recent advances in molecular genetic techniques have enabled us to understand more about the molecular basis of several genetic syndromes associated with mental retardation. In contrast, where there is no discrete cause, the interplay of genetic and environmental influences remains poorly understood. This article presents a critical review of literature on genetics of mental retardation.Keywords: chromosomal abnormalities; genetic; mental retardation; X-linked Mental retardation is characterized by significantly below average intellectual functioning existing concurrently with related impaired limitations in two or more of the following applicable adaptive skills areas: communication, self-care, home living, social skills, community use, self-direction, health and safety, functional academics, leisure and work, manifest before the age of 18.[1] The term mental retardation is not in itself a diagnosis, as it does not inform about aetiology, prognosis, or specific treatments. Rather, it refers to a clinical state that is developmental in origin and which affects intellectual and social functioning. Early literature made a distinction between ′pathological′ severe mental retardation (SMR) and ′familial′ (usually mild) mental retardation. More recently it has been customary to divide it into two groups according to IQ. Those who show IQ scores between 50 and 70 are categorized as having mild mental retardation (MMR) and those with IQ scores of below 50 are considered as having moderate-severe mental retardation (SMR).′ The impact of molecular genetics on our understanding of disease processes has been enormous and is likely to increase even further. The resurgence of interest in phenotypes combined with the new genetic techniques has reduced the proportion of those with moderate/severe mental retardation in whom the cause is unknown to 20%. Genetic causes may be hereditary or nonhereditary and may not produce specific syndromes. Over 500 recognized syndromes involving a genetic disorder have now been isolated and many have behavioural epiphenomena.[2] A specific cause for mental retardation can be identified in approximately 80% of people with SMR (IQ<50) and 50% of people with MMR (IQ 50-70). In this article, we will begin by considering what is known about the genetics of idiopathic mental retardation and then move onto discuss specific genetic causes of mental retardation. Search methodology Idiopathic mental retardation Idiopathic mental retardation refers to individuals who show no evidence of gross chromosomal defects or single-gene anomalies. It is sometimes considered as representing the lower end of the IQ distribution. IQ scores have been shown to have an average weighted correlation of 0.86 for monozygotic twins and 0.61 for dizygotic twins and an average correlation between first-degree relatives of 0.4, suggesting an overall heritability of 50%.[3] A recent study found twin concordances for mild mental impairment was 74% for monozygotic twins, 45% for same-sex and 36% for opposite sex dizygotic twins with a group heritability of 0.49.[4] The aetiology of idiopathic mental retardation is usually explained in terms of the ′polygenic multifactorial model.′ The difficulty is that there are too few studies to provide sufficiently precise estimates of the likely role of genes and environment in determining idiopathic mental retardation. Given the dearth of published literature we are reliant on attempting to draw conclusions from family and twin studies, most of which are very old and which report widely varying recurrence risks.[5] Recent studies of MMR have shown high rates of chromosomal abnormalities and this raises the possibility that a proportion of individuals with idiopathic mental retardation may have undetected or unknown chromosomal aberrations or single-gene defects. Further advances in molecular genetics may find idiopathic mental retardation to be an aetiologically heterogeneous group with some individuals showing retardation secondary to specific genetic causes, others because of environmental effects and the remainder due to multifactorial causes.Single-gene defects Single-gene defects account for only a small proportion of mental retardation and are more likely to be seen in individuals with SMR. These are well-recognized Mendelian conditions and are characterized by autosomal recessive, autosomal dominant or X-linked patterns of inheritance. Many single-gene disorders are syndromic. Syndrome recognition facilitates diagnosis and has allowed discrete phenotypes to be delineated. It may be aided by the reference to a dysmorphology database such as the London Dysmorphology Database-LDDB (http://dhmhd.mdx.ac.uk LDDB.html). The second database is the Online Mendelian Inheritance in Man-OMIM (http://www.ncbi.nlm.nih.gov/Omim/). Another large group of single-gene disorders causing mental retardation is the in-born errors of metabolism, which have been well documented by Scriver et al.[6] In some circumstances, different mutations occurring within the same gene may cause different clinical phenotypes, depending on how the mutation affects the production and function of protein involved. This phenomenon is known as allelic heterogeneity. The majority of inborn errors of metabolism follow an autosomal recessive pattern of inheritance, with some notable exceptions, e.g. Hunter syndrome. [Table - 1] describes some of the common single-gene defect disorders causing mental retardation. X-linked mental retardation Fragile X syndrome Fragile X syndrome is the most common inherited cause of mental retardation and originally derived its name from the characteristic nonstaining band or fragile site on the X-chromosome. It affects approximately 1 in 4425 to 6045 males and causes mental retardation in 1 in 8000 females. This syndrome accounts for half of all the X-linked mental retardation cases and around 0.6% of the population who show mental retardation. The mean IQ scores in Fragile X males seems to decline with increasing age, a phenomenon also described in people with Down syndrome.[11] A curvilinear relationship exists between the length of CGG repeat (the mutation) and the level of intelligence in Fragile X adults.[12] In normal individuals between 6 and 54 CGG repeats are expected with an average of 30 repeats. The mutation for Fragile X is a heritable unstable sequence of trinucleotide CGG repeats ranging from 230 to over 1000.[13],[14] The full mutation, when the repeat sequence reaches a critical length of about 200 copies, is associated with hypermethylation of the repeat and adjacent region. This results in the failure of FMR1 transcription and an absence of the FMR1 gene protein product (FMRP), which is responsible for the characteristic clinical features of fragile X syndrome.[15] ′Anticipation′occurs when premutations often expand to full mutations while transmitted by female carriers and the clinical severity of the disease increases with each successive generation. A milder form of mental retardation expressed as FRAXE occurs by FMR-2 mutation.[16] The key clinical characteristics of fragile X syndrome are mental retardation, large ears and a long face and macro-orchidism. In some adults, there is a characteristic facial appearance, with a large forehead with supra orbital fullness, long face, long nose, prominent jaws, high-arched palate and large ears with a bat-eared appearance. Eye abnormalities such as pale irises may be a subtle finding in some cases.[17] Epilepsy is reported in about 25% of the cases. Many affected individuals show higher rates of speech and language problems, attentional difficulties and hyperactivity and autistic-like features such as gaze avoidance and hand flapping.[18],[19] The observation of this type of behavioural phenotype in conjunction with early reports of increased rates of Fragile X in autism led to interest in the relationship between Fragile X and autism. Chromosomal abnormalities Chromosomal abnormalities account for 35-40% of SMR and 10% of MMR.[20],[21] Chromosomal aneusomies (loss or excess of chromosomal material) cause a gene dosage difference for a large number of genes and the phenotypic effect is pleiotropic; therefore, they always cause syndromes of multiple congenital anomalies and mental retardation.[22] The commonest autosomal abnormalities are trisomies, particularly involving chromosomes 13, 18 and 21 and these are all associated with increased maternal age. The most common aneusomy in live newborns is trisomy 21 (Down syndrome), which is invariably associated with mental retardation. Almost all chromosomal aneuploidies, which involve an alteration in the amount of chromosome material, are associated with mental retardation. It is not certain whether this is due to dosage effects of specific genes within the duplicated or deleted segments, or to a more general effect of aneuploidy per se. It is certainly true that individuals with chromosomal aneuploidy share some nonspecific features in common such as poor growth, microcephaly, epicanthic folds and unusual palmar creases, in addition to features more specific to the chromosomes involved. Some chromosome abnormalities occur in the mosaic form (where some cells show the normal 46 chromosomes and others have an extra chromosome) and for disorders, which are usually seen in the full form, mosaicism will confer a milder phenotype. However, there are some conditions, which are lethal in the full form and are therefore found only in the mosaic form in surviving individuals. The common chromosomal abnormalities associated with mental retardation are summarized in [Table - 3]. Down syndrome It is the most common cause of mental retardation and affects approximately 1 in 1000 live births. The incidence rises with advancing maternal age at the time of conception. Approximately 94% of the cases are caused by trisomy 21, 3.5% by translocation and 2.5% by mosaicism. The majority of individuals have moderate to SMR and some of those with a milder phenotype have been shown to have a mosaic karyotype. The critical region for the Down syndrome phenotype is in the region of bands 21q21.3-21q22.[23] Those with Down syndrome have characteristic clinical features including short stature, round skull, brachycephaly, neonatal hypotonia, a flat occiput, flat facial profile, small simply formed ears, protrusion of the tongue, high arched palate and typical eye signs (e.g. mongoloid slope of the palpebral fissures, epicanthic fold, Brushfield spots, premature cataracts, myopia, nystagmus, strabismus, etc.). Common abnormalities seen in the limbs include a single-palmar crease (simian crease), incurved fifth fingers, syndactyly (webbed fingers) and a wide gap between the first and second toes (sandal gap). Congenital heart defects (e.g. atrial or ventricular septal defect, mitral valve prolapse, patent ductus arteriosus) occur in 40% of infants and are a common cause of death. There is predisposition to Hirschprung′s disease, hypothyroidism and leukaemia and the early onset of Alzheimer′s disease. Microdeletion syndromes Recent advances in cytogenetic analysis techniques such as high resolution chromosome banding and fluorescent in situ hybridization (FISH) together with microsatellite analysis[24],[25] have enabled the detection of increasingly small chromosomal abnormalities. The microdeletions are usually small- (4 kb) or less- and encompass multiple genes, which may all contribute to the phenotype. Those microdeletions, which are observed most commonly, tend to have similar breakpoints, occurring in regions of the chromosome where there is a repetitive DNA sequence. Di George syndrome (velocardiofacial syndrome) Prader-Willi syndrome The region of chromosome 15q11-13 is subject to the phenomenon known as genomic imprinting. The genetic information of maternal and paternal origin is manifested differently in this region. The individuals who have a microdeletion of 15q11-13 manifest different characteristics depending on whether the deletion is maternal or paternal in origin. A paternal deletion gives rise to features of the Prader-Willi syndrome[29] and a maternal deletion gives rise to features of Angelman syndrome.[30] These conditions can also arise from maternal and paternal uniparental disomy for chromosome 15 leading to Prader-Willi syndrome and Angelman syndrome, respectively. Prader-Willi syndrome is also known as HHHO (H, hypotonia; H, hypogonadism; H, hypomentia; O, obesity). It is a rare condition with a prevalence of 1.2-1.3 per 10 000. It is characterized by neonatal hypotonia, hyperphagia (over eating), obesity, hypogonadism, short stature and mild to moderate mental retardation. Feeding difficulties such as absence of swallowing and sucking reflexes are common in the initial stages. The hands and feet are often small and the facies are distinctive, with bitemporal narrowing and almond-shaped palpebral fissures. Abnormalities in speech, sleep and behaviour are common. A remarkable area of cognitive strength is their visual-spatial integration as they may show unusual skill with jigsaw puzzles.[31] Temper tantrums, self-injurious behaviour particularly in the form of skin picking and obsessional behaviour are frequently observed.[32] Angelman syndrome A deletion in the maternally derived long arm of chromosome 15 (q11-q13) will give rise to the symptoms of Angelman syndrome. It is also known as the ′happy puppet syndrome′.[30] It affects between 1 in 20 000 and 1 in 30 000 people and is characterized by ataxia, jerky limb movements and abnormal gait, absent speech and SMR. Typical facial features consist of a long face and prominent jaw, a wide mouth with widely spaced teeth, thin upper lip, mid facial hypoplasia, deep set blue eyes, blonde hair and microcephaly. Other characteristic features include epilepsy (about 86%) and/or an abnormal EEG, inappropriate bouts of laughter, tongue thrusting movement, mouthing behaviour and a happy, social disposition.Sex chromosome anomalies Abnormalities in the number of sex chromosomes are generally less devastating in their effects than aneuploidies in the autosomes. Sex chromosome anomalies occur in roughly 1 in 400 live births.[33] These abnormalities cause mild decreases in IQ (5-15 points) in relation to the family′s mean IQ. These anomalies are commonly due to chromosomal nondisjunction, the risk of which increases with maternal age. The most common include Turner′s syndrome (45,XO), Klinefelter′s syndrome (47XXY) and the 47XXX and 47 XYY karyotypes. All four are associated with a slight decrease in IQ, as evidenced by comparison with siblings and by their higher frequency in mentally retarded populations.[34]Turner′s syndrome Turner′s syndrome affects 1 in 2000 to 1 in 5000 females. It is characterized by the complete or partial absence of one X-chromosome (45XO). The clinical features include short stature, a webbed neck, increased carrying angle of the elbow, failure to develop secondary sexual characteristics and visuo-spatial deficits. Interesting recent data suggest the X-chromosome might also play an important role for other sorts of cognitive skills, which facilitate social interaction. Girls tend to show superior levels of skills such as the ability to respond to cues in the behaviour of others, to inhibit distractions and to develop strategies of action compared with boys. Females affected by Turner′s syndrome, whose X-chromosome is of maternal origin show poorer social cognitive skills than those who possess a paternally derived X-chromosome.[35]Klinefelter′s syndrome Klinefelter′s syndrome (47XXY) affects approximately 1 in 1000 males. In about two-thirds of cases the anomaly is due to maternal nondisjunction and is nonfamilial.[36] These individuals have high rates of speech and language disorders (Ratcliffe 1994). It was originally thought that affected individuals showed increased rates of mental retardation, psychiatric disorder and criminality. However early studies of the XXY behavioural phenotype were based on highly selected samples of those who had been institutionalised,[37] and it is now recognized that the majority who possess this karyotype are of normal intelligence and show no increased rates of behavioural psychiatric difficulties.[38],[39]Conclusions Recent advances in identifying previously undetected sub-microscopic chromosomal anomalies have proven to be successful. They highlight the fact that many cases of idiopathic mental retardation may have specific aetiologies, which may not yet be discovered. Given the increased interest in studying behavioural phenotypes associated with some of these conditions, it is hoped that the localization of specific genes and the identification of genes within critical chromosomal regions might pave the way for understanding more about the genetic basis of behaviour and psychiatric disorders. The challenge for the future is to unravel the complex interaction between genes and environment using well-designed twin and adoption studies as well as molecular genetic strategies so as to determine the extent to which genes and environment contribute, co-act and interact in determining intelligence. Such research will contribute in planning appropriate interventions. REFERENCES
Copyright 2005 - Indian Journal of Medical Sciences The following images related to this document are available:Photo images[ms05063t2.jpg] [ms05063t1.jpg] [ms05063t3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}