 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
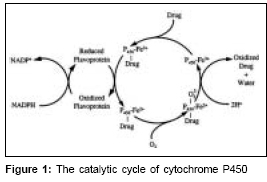
Indian Journal of Medical Sciences, Vol. 61, No. 2, February, 2007, pp. 102-116 Practitioners section Cytochrome P450 enzyme isoforms and their therapeutic implications: An update Kalra BhupinderSingh Department of Pharmacology, Maulana Azad Medical College, Bahadur Shah Zafar Marg, New Delhi - 110 002 Code Number: ms07017 Abstract Clinicians should be cognizant of potential drug drug interactions and become familiar with the substrates, inhibitors and inducers of the common enzymatic pathways responsible for drug metabolism. Our knowledge of and ability to predict drug interactions have improved with growing understanding of substrates, inhibitors and inducers of cytochrome P450 (CYP-450) isoenzymes. These isoenzymes are a major determinant of the pharmacokinetic behavior of numerous drugs. In addition to inhibition and induction, microsomal drug metabolism is affected by genetic polymorphisms, age, nutrition, hepatic disease and endogenous chemicals. Prescribing physicians by understanding the unique characteristics of these isoenymes may better anticipate and manage drug drug interactions.Keywords: Cytochrome P450, drug interaction, enzyme inhibitor Introduction Biotransformation or metabolism of drugs leads to chemical alteration of the drug in the body. It is needed to render non polar (lipid soluble) compounds polar (lipid insoluble) so that they are not reabsorbed in the kidney tubules and are excreted. Biotransformation reactions can be classified as either Phase 1 functionalization reactions or Phase 2 biosynthetic (conjugation) reactions. Phase 1 reactions introduce or expose a functional group (-OH, -NH 2 , -SH) on the parent compound, metabolite formed are inactive but in some instances active metabolites are also formed. Phase 1 reactions generally lead to loss of pharmacological activity e.g., morphine, propanolol, pentobarbitone etc. Prodrugs (pharmacologically inactive compounds but after metabolism have active metabolite) gets bioactivated after phase 1 reactions e.g., levodopa, enalapril etc. Phase 2 conjugation reactions lead to the formation of a covalent linkage between a functional group on the parent compound or phase 1 metabolite with endogenously derived glucuronic acid, sulfate, glutathione, amino acids or acetate. These highly polar conjugates are generally inactive and are excreted rapidly in urine and feces. The enzyme involved in phase 1 reactions are located primarily in endoplasmic reticulum, while the phase 2 conjugation enzyme systems are primarily cytosolic. Metabolites can alter the pharmacological action of drug qualitatively. Terfenadine, a non sedating antihistaminic, can, rarely cause serious cardiac dysrythmias by blocking cardiac potassium channels whereas its pharmacologically active metabolite (fexofenadine) blocks histamine H 1 receptors but not cardiac potassium channels and it has replaced terfenadine. The selective serotonin reuptake inhibitors (SSRIs) like fluoxetine is demethylated to norfluoxetine which is pharmacologically active and has a elimination half life of 10 days as compared to parent compound which is just 50 hrs. Norfluoxetine also competes with other agents for hepatic oxidases to elevate plasma concentrations of other agents, including tricyclic antidepressants, days after administration of the parent drug has been stopped. Cytochrome P450 isoenzymes (CYPs) The Cytochrome P450 isoenzymes (CYPs) are superfamily of haemoprotein enzymes found on the membrane of endoplasmic reticulum. They are responsible for catalyzing the metabolism of large number of endogenous and exogenous compounds. CYPs are also known as mixed function oxidases and mono-oxygenases as metabolism of a substrate by a CYP consumes one molecule of molecular oxygen and produces an oxidized substrate and another molecule of oxygen appears in water as byproduct.[1] CYPs are also called polysubstrate mono-oxygenases as one isoenzyme can have multiple substrates.[1],[2] These enzymes are responsible for biotransformation of drugs and are body′s defense against xenobiotics along with P-glycoprotein. P-glycoprotein is efflux pump or transporter present in brain capillary endothelial cells, intestinal mucosal, renal and tubular cells, hepatic canalicular cells etc and are responsible for extrusion or efflux of drugs thereby enhancing drug elimination. Cytochrome P450 isoenzymes are predominantly present in liver but are also found in intestine, lungs, kidneys, brain etc. Biotransformation of drugs by these enzymes render them ionic and more water soluble so that they can be excreted, drawback of this process is limited bioavailability of drugs.[3] CYPs catalyse variety of reactions including N-dealkylation, O-dealkylation, S-oxidation, epoxidation and hydroxylation. A typical CY P450 catalysed reaction: NADPH + H+ + O 2 + RH → NADP+ + H 2 O + R-OH RH denotes parent drug and R-OH is oxidized product[4] [Figure - 1]. Other oxidative enzymes such as dehydrogenases and flavin containing monooxygenases (FMOs) also are capable of catalyzing the metabolism of specific drugs, but, in general such enzymes are of minor overall importance. Cytochrome P450 isoenzymes are designated by the letters e.g., CYP2D 6 , "CYP followed by an Arabic numeral, a letter and another Arabic numeral (italics). ′P′ in cytochrome P450 stands for ′pigment′.[5] Each enzyme is termed as isoform since each is derived from a different gene. These isoforms have a spectrophotometric absorption peak at or near 450 nm when bound and reduced by carbon monoxide.[5],[6] Cytochrome P450 isoenzymes nomenclature based on the presence of common amino acid sequence was proposed by Nebert and colleagues;[7] Families - proteins with at least 40% amino acid sequence homology. In humans upto 21 families have been described. e.g., CYP2, CYP3. Subfamilies - members of the same family must have at least 55% amino acid sequence homology. e.g., CYP2D, CYP3A. Individual genes - they are denoted by Arabic numeral (italics), there are 50 or so genes important in man. e.g., CYP2D 6 , CYP3A 4. In humans upto 21 families, 20 subfamilies and 57 genes have been described. Out of these CYP 1, 2 and 3 account for 70% of total hepatic CYPs content and are responsible for 94% of drugs metabolism in liver.[8] To identify which P450 isozymes are responsible for metabolizing drugs, several in vitro approaches have been developed. Some of these approaches include: (a) metabolism by microsome derived from cDNA-expressed enzyme, (b) use of selective inhibitors with microsomes, (c) Immunoinhibition of CYP by isoforms specific anti-P450 antibodies in microsomes and (d) correlation of drug candidate metabolites formation with several isoform specific P450 activities in a panel of liver microsomes.[9] A combination of approaches is typically required to accurately identify which P450 isoenzyme is responsible for metabolizing a drug. However, the use of cDNA expressed P450s for the preliminary determination of the principal CYPs involved in a drug candidate′s metabolism is a reasonable starting point in a drug discovery setting. Drug interactions involving the CYP isoforms generally result from one or two processes, enzyme induction or enzyme inhibition. Enzyme inhibition usually involves competition from another drug for enzyme binding site. This process usually begins with the first dose of the CYP inhibitor. Thus, this review is an attempt to briefly summarize the current understanding of CYP isoforms and various drug interactions resulting from drugs metabolized by these isoenzymes, also this review provides hands on information on drug interactions faced by clinicians in routine practice. Clinically important aspects of CYP450 drug metabolism:
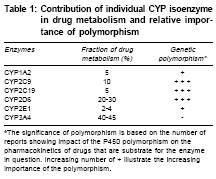
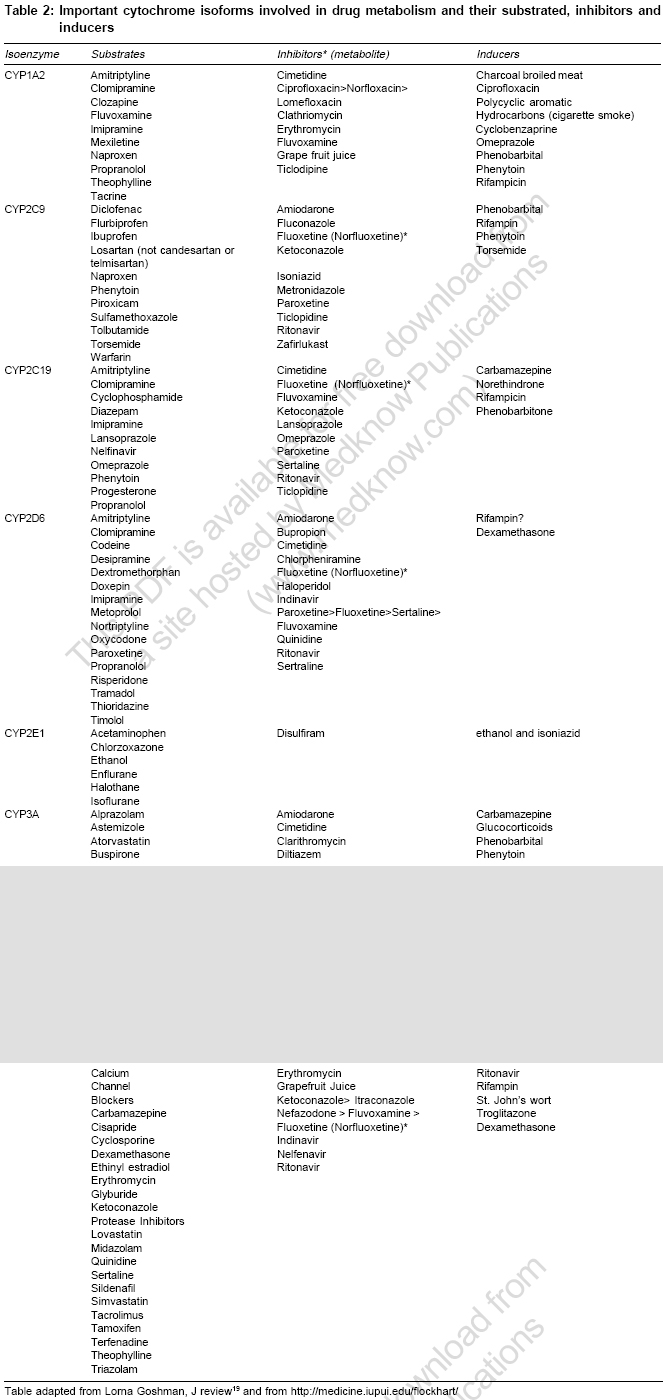
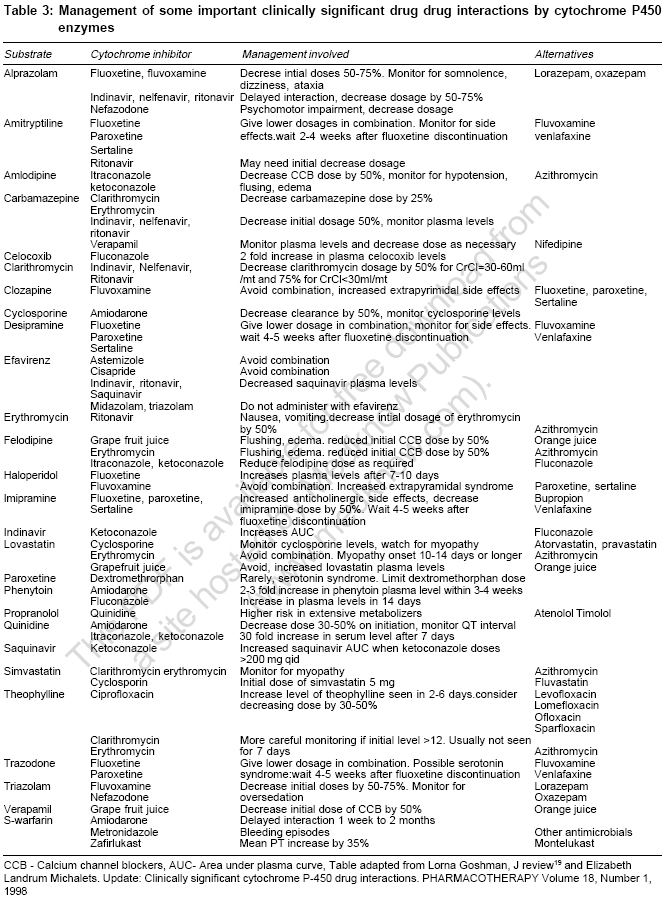
Genetic polymorphism In different population and individuals activity of CYPs differs. Genetic variations in a population is termed as ′polymorphism′ when both gene or allelic variants exist with a frequency of at least one percent. Individual′s genetic inheritance do affects the body′s response to drugs. It may be possible to predict therapeutic failures or severe adverse drug reactions in individual patients by testing for important DNA sequence variations or polymorphisms (genotyping) in key drug-metabolizing enzymes, receptors, transporters, etc. Potentially, test results could be used to optimize drug choice, avoid serious adverse effects and decrease medical costs.[9] In 1978, the first CYP450 was purified to homogeneity, which is now termed as CYP2B4.[10] Three years later in 1981, Fujii-Kuriyama presented the first cDNA sequence in 5th Microsomes and Drug oxidation meeting in Tokyo.[10] At that time it was difficult to foresee genetic variants of CYPs that can cause inter individual variability in drug response and can also lead to adverse drug reactions (ADRs). All genes coding CYP enzymes in families 1-3 are polymorphic. The polymorphic forms are responsible for development of significant number of ADRs. It has been estimated that 56% of drugs reported in ADRs studies are metabolized by polymorphic phase 1 enzymes, of which 86% are CYPs. CYP2C9, CYP2C19 and CYP2D6 are the main polymorphic forms responsible for almost 40% of P450 mediated drug metabolism[10] [Table - 1]. CYP3A 4 is highly conserved across different individuals and essentially no functionally variant forms have been observed in orientals and Caucasians. But still CYP3A activity shows marked interindividual variability (>10 fold), this property is suggested to be of genetic origin although the reasons for this have not been explained. Major allelic variants of P450 genes of clinical importance have been identified. Based on allelic variants, four phenotypes have been identified: Poor metabolizers which lack functional enzymes, intermediate metabolizers who are heterozygous for one deficient allele, extensive metabolizers who have 2 normal alleles and ultrarapid metabolizers who have multiple gene copies.[10] Ultrarapid metabolizers of an active drug may not reach therapeutic concentrations at usual recommended doses of active drugs due to increase in enzyme activity, while poor metabolizers may suffer more adverse events at usual doses due to reduced enzyme activity leading to increased concentrations of the drug. Conversely, for administered prodrugs that must be bioactivated by CYP450 enzymes into active metabolites, ultrarapid metabolizers may suffer adverse effects and poor metabolizers may not respond. All the major human drug-metabolizing P450 enzymes have been identified and cloned and the major gene variants that cause inter-individual variability in drug response and are related to adverse drug reactions have been identified. This information now provides the basis for the use of predictive pharmacogenetics to yield drug therapies that are more efficient and safer. It has been observed that patients on antipsycotics develop parkinsonism like side effects with high frequency in poor metabolizers compared with extensive metabolizers. Oversedation has been observed in poor metabolizers in many studies following treatment with thioridazine and other antipsycotics. Polymorphic CYP2C9 accounts for majority of phenytoin metabolism, several side effects have been reported including CNS intoxication such as ataxia, diplopia in patients with defective CYP2C9 alleles following phenytoin treatment.[11] It is anticipated that personalized or tailor made treatment will be offered in the future based on genotype of individual in vast number of subjects, decreasing the frequency of ADRs and cost incurred on treating ADRs. Major pharmaceutical companies do take pharmacogenetic aspect of CYP450 into account during drug development and any compound showing high affinity for a polymorphic P450 enzyme is terminated if pharmalogically competitive candidates are not substrate for the polymorphic enzyme one at hand.[12] Recently, the AmpliChip (Roche Molecular Systems, Inc.) was cleared for marketing by the FDA. The AmpliChip is a microarray consisting of many DNA sequences complementary to tw°CYP450 genes applied in microscopic quantities at ordered locations on a solid surface (chip). The AmpliChip tests the DNA from a patient′s white blood cells collected in a standard anticoagulated blood sample for 29 polymorphisms and mutations for the CYP2D6 gene and two polymorphisms for the CYP2C19 gene.[13] According to FDA labeling, "Information about CYP2D6 genotype may be used as an aid to clinicians in determining therapeutic strategy and treatment doses for therapeutics that are metabolized by the CYP2D6 product".[13] Drug Drug interaction One of the predominant reason for drug drug interaction is overlapping substrate specificities by the CYPs. When two co administered drugs are both metabolized by a single CYP, they compete for binding to enzyme′s active site. This can result in inhibition of metabolism of one or both the drugs, leading to elevated plasma levels. If there is narrow therapeutic index for the drugs, the elevated serum levels may elicit unwanted toxicities. Similarly, the same drug can be metabolized by more than one CYP isoform and that the relative contribution of a specific enzyme for the total elimination of a drug determines the degree to which a pharmacokinetic interaction resulting from inhibition of that enzyme can occur. Drug Drug interations are the leading cause of ADRs. It has been estimated that ADRs cause > 100,000 deaths annually in US and upto 7% of all hospital admissions in the UK and Sweden are due to ADRs.[14] Since most of drug drug interactions are due t°CYPs, it becomes important to determine the identity of the CYP that metabolizes a particular drug and to avoid co administering drugs that are metabolized by the same isoenzyme. Enzyme induction On repeated administration drugs can induce CYP450 by enhancing the rate of its synthesis or reducing its rate of degradation but mainly in induction absolute increase in enzyme synthesis is seen. This induction leads to increase in rate of metabolite production, increased hepatic biotransformation, decrease in serum half life of drug, shorten drug response and could also lead to pharmacokinetic tolerance.[15],[16] Xenobiotic influences the extent of drug metabolism by activating transcription and inducing the expression of genes encoding drug metabolism enzymes.[16],[17],[18] The mechanism of induction is incompletely understood but is similar to that involved in action of steroid that bind to nuclear receptors. Drugs bind to the ligand binding domain of a soluble protein, termed the aromatic hydrocarbon (Ah) receptor which is cytosolic in nature. This binding leads to conformational change in receptor which facilitates transportation of this complex to the nucleus by an Ah-receptor nuclear translocater and binds Ah-receptor response elements in the DNA thereby promoting transcription of the gene. For example, inducible expression of CYP3A is regulated by ligand-activated nuclear receptors CAR (constitutively active receptor) and can be activated by a wide variety of antibiotics, barbiturates etc. A similar kind of receptor mechanism invoving the pregnane X receptor (PXR) is also involved in the induction of CYP3A. In the case of CYP1A isoform, induction is through the binding of a ligand to Ah-receptor. Although CYP induction is normally the consequence of an increase in gene transcription, some non-transcriptional mechanisms also have been reported e.g., induction of human CYP2E 1 by ethanol and isoniazid is not transcriptional, but it results from protein stabilization or increased protein translation.[9] The onset and duration of induction depends both on the kinetics of the drug and half life of CYP enzyme, which ranges from 1-6 days.[16] Usually it takes 4-14 days for peak induction and on withdrawing inducer the CYP returns to their original level in 1-3 weeks.[19] Phenobarbital induce changes may be delayed for 14-22 days because of the long interval (long t 1/2 ) before peak phenobarbital plasma levels are reached. In contrast, the onset of rifampicin induced increase in enzyme activity is only 4 days. 20-40 fold increase in enzyme activity is seen with phenobarbital whereas alcohol increase enzyme synthesis by 1-2 fold. Induction of CYP3A by rifampicin can lead to decrease in AUC of saquinavir by 80% and of ritonavir by 35%.[16] In addition to phenobarbital and rifampicin, glucocorticoids, macrolides, anticonvulsants induce specific forms of CYP3A. Isoniazid and chronic alcohol intake induce CYP2E1 which oxidizes ethanol and activates carcinogenic nitrosamine.[15] It must also be noted that an inducer may enhance not only the metabolism of other drugs but also its own metabolism. Thus, continued use of some drugs may result in a pharmacokinetic type of tolerance. Important non pharmacological inducers include polycyclic aromatic hydrocarbons in cigarette smoke and charcoal broiled meat.[20] They causes induction of CYP1A2 and results in higher dosing requirements of drugs like theophylline in smokers. Other environmental chemical known to induce specific isoenzyme include polychlorinated biphenyls, which are used widely in industry as insulating materials and plasticizers. It has been observed that with age inducibilty of CYPs decreases.[21] Enzyme inhibiton By identifying the isoenzyme that metabolizes the drugs, some potential drug-drug interaction can be predicted, often because inhibition is specific. For example, inhibition of metabolism of tricyclic antidepressants like desipramine by fluoxetine.[22] Desipramine is metabolized by CYP2D6 and its metabolism is strongly inhibited by binding of fluoxetine to the same isoenzyme. The duration of interaction depends upon half life of drug. Enzyme inhibition leads to decrease in rate of hepatic biotransformation of the drugs causing increased serum concentration and toxicity.[20] Inhibition of CYP enzymes can be classified grossly into reversible inhibition and irreversible inhibition based on the enzymatic mechanism.[12] Reversible inhibition is the most common mechanism in documented drug-drug interaction cases. Reversible inhibition occurs as a result of direct competition for the binding site on a CYP enzyme between a substrate and an inhibitor. The competition can be either for the heme prosthetic group or for other regions of the active site. Compounds with lone electron pairs tend to be potential reversible CYP inhibitors because the haem prosthetic group is the oxidation center for CYP-catalyzed reactions. The determinant of potency of an inhibitor is the strength of the bond between the prosthetic heme iron and the lone electron pair of inhibitors. For example, cimetidine, an H2-receptor antagonist, is used in the treatment of gastric ulcers. The biological activity appears to involve the imidazole ring of cimetidine with competitive binding to the H 2 receptor. However, the imidazole ring of cimetidine also binds to the haem prosthetic group and exhibits selective inhibition of biotransformation reactions catalyzed by CYP3A4 and 2D6. ketoconazole and cimetidine are both imidazole-containing compounds and CYP3A4 inhibitors, but ketoconazole is a more potent CYP inhibitor than cimetidine probably as ketoconazole has a higher lipophilicity that results in stronger hydrophobic interactions t°CYP3A4 compared to cimetidine with a lower lipophilicity.[12] In contrast to the reversible inhibition in which two agents compete for the active sites on the enzyme, irreversible inhibition is caused by reactive metabolites generated from CYP-catalyzed reactions. The first type of irreversible inhibition involves the formation of metabolic intermediate (MI) complexes. Inhibition of CYP3A by erythromycin is a well documented example that results from a metabolic intermediate complex.[12] Erythromycin contains a tertiary amine in the amino sugar ring. Transformation reactions, such as N-hydroxylation, N-demethylation and N oxidation catalyzed by CYP3A, generates a nitroso metabolite that binds tightly to the haem portion of the CYP enzyme to form a stable MI complex. The MI complex is inactive. As a potent antimicrobial agent, erythromycin inhibits a number of drug oxidation reactions catalyzed by CYP3A4.[23],[24],[25] It inhibits the oxidation of terfenadine, cyclosporin and numerous other drugs both in vitro and in vivo .[26] CYP1A 2 CYP1A2 accounts for 13% of total hepatic content of isoenzymes.[8] CYP1A2 isoenzyme metabolizes chemicals and environmental toxins. CYP1A2 can activate benzpyrene, a carcinogen present in cigarette smoke, by 7-8 epoxidation.[20] Cigarette smoke can increase the synthesis of this isoenzyme by 3 fold. Drug substrates include caffeine, propranolol and theophylline etc. Drugs that inhibit CYP1A2 include cimetidine, erythromycin, floroquinolones. Out of floroquinolones, ciprofloxacin is the most potent inhibitor as compared to norfloxacin, ofloxacin. Sparfloxacin and levofloxacin have negligible effect on enzyme activity[27] [Table - 2]. CYP2C 9 CYP2C9 exhibits genetic polymorphism [Table - 1]. CYP2C9 is absent in about 1% of Caucasians. CYP2C9 is responsible for the metabolism of ibuprofen, tolbutamide and torsemide.[28],[29],[30] S-warfarin is mainly metabolized by this isoform along with various NSAIDs.[31] Inhibition of this isoform results in several clinically important drug interactions. Fluconazole, metronidazole and amiodarone are a few examples of the many drugs that profoundly inhibit S-warfarin metabolism and produce marked increase in prothrombin time [Table - 2]. Other serious interaction is inhibition of S-warfarin metabolism by amiodarone.[32] Because of long half life of amiodarone, clinical onset of interaction may be delayed by one week to 2 months and PT remains elevated for 1-3 week after treatment is discontinued. This interaction can be avoided by decreasing the dose of warfarin by 25% when amiodarone is added. Induction of CYP2C9 by rifampicin can cause therapeutic failure of phenytoin [Table - 3]. CYP2C 19 CYP2C19 also exhibit polymorphism. About 20% of Asians and 3-5% of Caucasians are poor metabolizers, whereas in 20% of Japanese this isoform is absent.[8] Drug metabolized by CYP2C19 includes omeprazole, diazepam, lansoprazole. Poor metabolizers taking imipramine, clomipramine or diazepam may develop higher serum levels than extreme metabolizers. CYP2D 6 This isoform accounts for 2% of total CYP450 expression and exhibit genetic polymorphism. 7-10% of Caucasians are poor metabolizers.[10] 5-10% Mexican-Americans lack CYP2D6 isoenzyme but only 1-2% of Asians lack this enzyme and are characterized as poor metabolizers.[10],[11] The activity of CYP2D6 shows a bimodal distribution indicating the presence of dominant and recessive alleles in the population. CYP2D6 is not inducible by pharmacological agents. More than 80 drugs in clinical use are metabolized by this isoform. Several antipsychotic are metabolized by CYP2D6, so poor metabolizers are at risk for postural hypotension and extrapyramidal side effects. Codeine is O-demethylated to morphine by CYP2D6.[33] Thus, poor metabolizers may have less response to codeine as compared to extensive metabolizers. All antidepressants except fluvoxamine, nefazodone, bupropion and citalopram either inhibit or are metabolized by CYP2D6. Paroxetine is most potent in inhibiting the metabolism of CYP2D6 substrates as compared to sertaline and fluoxetine. Inhibition of CYP2D6 by fluoxetine or paroxetine can increase the cardiac toxicity of tricyclics if both SSRIs and tricyclics are administered together.[34] Fluoxetine also can inhibit metabolism of trazodone and nefazodone to precipitate serotonin syndrome.[35] CYP2E 1 CYP2E1 accounts for 7% of total CYP content in liver. It exhibit polymorphism and toxicologically an important enzyme. Several volatile anesthetic agents (sevoflurane, isoflurane) are metabolized by CYP2E1.[36] This isoform is also responsible for metabolism of ethanol and acetaminophen, chronic ethanol consumption induces but acute administration of ethanol inhibits CYP2E1. Patient with alcohol dependence are at increase risk for acetaminophen toxicity as ethanol induction of CYP2E1 increases formation of reactive metabolite which is hepatotoxic.[37],[38] Cimetidine exhibit only moderate affinity for this isoform and produces no significant inhibition of the production of acetaminophen′s toxic metabolites.[39] CYP3A 4 CYP3A4 family accounts for 30% of total hepatic content and 70% of gut wall content.[8] It does not exhibit genetic polymorphism. CYP3A4 is involved in metabolism of large number of endogenous and exogenous compounds. The endogenous compounds metabolized by CYP3A4 includes progesterone, oestradiol, testosterone and cortisol.[40] Some of the most serious drug interactions have been caused by accumulation of substrate metabolized by CYP3A4 like astemizole, terfenadine. Potent inhibitors of this isoform like erythromycin, ketoconazole and grape fruit juice have caused elevated substrate levels that have precipitated prolonged QT interval, torsades de pointes and even death.[41] Grape fruit juice causes downregulation of CYP3A4 production in small intestine which can lead to increase bioavailability of felodipine, cyclosporine, saquinavir, terfenadine and triazolam.[41],[42] Serious ventricular arrhythmias have been reported in patients taking cisapride and drugs that inhibit CYP3A4, the isoform responsible for metabolism of cisapride.[43] Erythromycin and clarithromycin (but not azithromycin) decrease theophylline metabolism by inhibiting CYP3A4 [Table - 2]. Rifampicin is very potent inducer of CYP3A4. Of particular clinical relevance is the potential reduction of oral contraceptive efficacy by rifampicin, since estradiol levels can be reduced by rifampicin mediated CYP3A4 induction. Rifampicin is also responsible for producing sub therapeutic protease inhibitors levels[44] [Table - 3]. Intestinal CYP3A4-mediated biotransformation and active efflux of absorbed drug by P-glycoprotein are major determinants of bioavailability of orally administered drugs. The evidence for hypothesis that CYP3A4 and P-glycoprotein (P-gp) may act in concert to limit oral drug bioavailability comes mainly from a limited number of in vitro and animal studies. CYP3A4 and P-gp are an integral part of an intestinal defense system to protect the body against xenobiotics and drugs that are substrates of both proteins often have a low bioavailability after oral administration.[45],[46] Therefore, like CPY-mediated drug interactions, P-gp-mediated drug interactions may be anticipated when P-gp substrates and P-gp inhibitors or inducers are coadministrated. Because both proteins are expressed in enterocytes and hepatocytes and contribute to a major extent to first-pass elimination of many drugs, there is an overlapping of substrate specificities and inhibitor/inducers between CYP3A4 and P-gp; many drug interactions may involve both CYP3A4 and P-gp and it is difficult to distinguish the specific nature of drug interactions at present. Some important drug interactions websites http://www.hospitalist.net/highligh.htm http://home.earthlink.net/~cpardee/ http://medicine.iupui.edu/flockhart/ http://vm.cfsan.fda.gov/%7Elrd/fdinter.html http://www.erowid.org/psychoactives/pharmacology/pharmacology_enzymes1. Conclusion An understanding of unique function and characteristics of CYP enzymes is must for the physicians as they face various drug drug interactions related morbidity in their routine practice and are a part to it too. Here, comes the role of clinical pharmacologists to provide clinicians with updated information regarding drug metabolizing enzymes and drug interactions caused by them in easy to comprehend(tabular, hand outs, booklets etc) forms so that they can better anticipate and manage drug drug interactions. References
Copyright 2007 - Indian Journal of Medical Sciences The following images related to this document are available:Photo images[ms07017t3.jpg] [ms07017f1.jpg] [ms07017t2.jpg] [ms07017t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}