 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
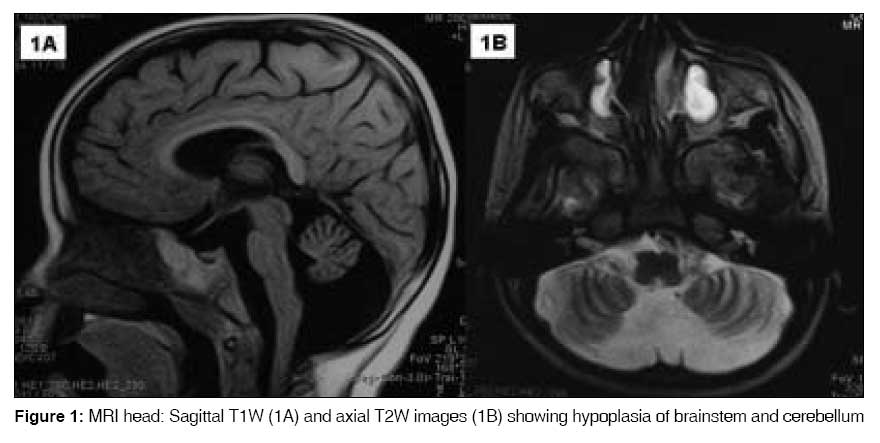
Indian Journal of Medical Sciences, Vol. 63, No. 3, March, 2009, pp. 120-123 Letter To Editor Macrocephaly in association with pontocerebellar hypoplasia type 1: A paradox Tamhankar ParagM, Suvarna JyotiC, Deshmukh ChandrahasT Department of Pediatrics, Seth GS Medical college and KEM Hospital, Mumbai, Maharashtra Code Number: ms09025 Sir, We recently diagnosed pontocerebellar hypoplasia type 1 in a 26-month-old male child who presented with regression of motor and mental milestones and progressive macrocephaly. We describe this unusual association, which has not been previously reported in literature. A 26-month-old male, second child of a farmer couple in a non-consanguineous marriage presented with seizures and loss of achieved milestones since ten months of age. Antenatal and birth history was normal. The birth weight was 2560 gm, birth length was 51 cm and head circumference was 34 cm. He had developed appropriate gross motor, socio-adaptive, and language milestones by ten months of age. He was noticed to be increasingly floppy after ten months. Parents felt that his head was "outgrowing" his body since he was about 15 months. He had cognitive decline and had lost his speech and parental recognition by 26 months. The couple′s first child had previously died at around 34 months of age from pneumonia and could not be examined by us. His available medical records suggested that he suffered from progressive motor and cognitive decline noted since one year of age. His head circumference was 35 cm (normal) at birth and 54 cm at 36 months prior to death (+ 3.4 SD for age). He was hypotonic and had depressed deep tendon reflexes. The child was not investigated. The present child examined by us at 26 months had a total length of 85 cm (10 th centile for age), head circumference of 52 cm (+ 3.5 SD for age) and weighed 11 kg (5 th centile). The child did not follow objects or light. He had a pendular nystagmus. The pupils were reacting to light. Fundus exam was normal. He had constant drooling of saliva and a weak gag. There was hypotonia in the limbs and axial musculature. Wasting was observed in the limb muscles, however power in all the limbs appeared to be more than 3/5 (Medical Research Council grading) with predominant distal weakness. Intermittent fasciculations were observed in limb muscles and the tongue. There was no exaggerated startle to loud sound. The deep tendon reflexes were depressed. Superficial reflexes were normal. His motor-adaptive DQ was 25 and cognitive DQ was 20. Examination of other systems did not reveal any abnormality. The head circumference of father was 54 cm and mother was 52 cm (appropriate for age and sex). MRI of brain revealed striking hypoplasia of the cerebellum and brainstem especially the pons. The cerebellum was small, the vermis was predominantly affected. The vermian folia and fissures were reduced in number but structurally normal (suggesting ′hypoplasia′ rather than ′atrophy′ which has shrunken folia and elongated fissures). [1] The lateral cerebellar hemispheres were moderately affected. The cerebral hemispheres appeared relatively atrophied; the cerebral and cerebellar gray and white matter did not show any abnormality in T2W and FLAIR images. The subdural spaces were increased [Figure 1A] and [Figure 1B]. Electromyography (EMG) revealed positive sharp waves and delayed recruitment. Fibrillation potentials were seen at rest. F waves were low in amplitude but latency was normal. Motor and sensory nerve conduction velocities were normal. Histopathological examination of the skeletal muscle showed increased variability in fiber diameter, some grouped atrophied fibers and predominance of type 1 fibers. These findings suggested chronic anterior horn cell degeneration. Total serum creatine kinase levels were 90 IU/L and MM fraction was 75 IU/L. Analysis of leukocyte enzymes hexosaminidase A and B, arylsulphatase A and galactocerebrosidase showed normal levels. There was no homozygous deletion of exons 7 and 8 of SMN1 gene. Electroencephalography, ultrasound abdomen, two-dimensional echocardiography, radiographs of the skull and the rest of the skeletal structure revealed no abnormality. Karyotype showed a normal male karyotype (46,XY) and he was cytogenetically negative for fragile X site. Urine and blood tested negative for abnormal metabolites on gas chromatography and tandem mass spectrometry. Pontocerebellar hypoplasia is diagnosed by a combination of pontine and cerebellar hypoplasia on neuroimaging. [1] The following six subtypes are described: type 1 including cases with anterior spinal motor neuron loss similar to Werdnig Hoffman disease; type 2 including cases with chorea/dystonia and not having spinal motor neuron loss; type -3 is similar to type 2 but with absence of dyskinesia, presence of optic atrophy. [2] Cases with congenital olivopontocerebellar hypoplasias (with C-shaped inferior olives) are classified as PCH-4 if vermis is spared or PCH-5 if vermis is not spared. [2] PCH type 6 presents as a fatal infantile encephalopathy. [2] Our case was labeled as pontocerebellar hypoplasia type 1 on the basis of the unique association of anterior horn cell disease with the characteristic MRI findings. PCH type 2, 3, 4, 5 and 6 were clinically ruled out. Congenital disorders of glycosylation (CDG) too can have PCH, but are accompanied by microcephaly, abnormal fat distribution, inverted nipples etc, which were absent in our case. CDG could not be ruled out due to non-availability of isoelectric focusing of serum transferrin. [2] Spinomuscular atrophy (SMA) was almost ruled out as about 95% of cases show deletion in exon 7 or 8 of SMN1 gene. Progressive cognitive decline is also not a feature of SMA. Another differential-infantile neuroaxonal dystrophy also presents with regression of milestones, pontocerebellar atrophy and similar EMG findings but they have spastic tetraplegia, MRI brain shows severe cerebral atrophy; cerebellar atrophy rather than hypoplasia; T2W and FLAIR images show cerebellar cortex and cerebral white matter hyperintensity; features that were absent in our case. [1] We speculate on the causes of progressive macrocephaly in our case. Brain atrophy in general is accompanied by microcephaly as brain growth is an important driver for skull growth. Hence progressive macrocephaly in association with pontocerebellar hypoplasia and cerebral atrophy (a neurodegenerative course) may seem paradoxical in our case. This was probably due to skull growth exceeding the brain growth rather than neuronal or glial proliferation as suggested by the increased subdural space and relative cerebral atrophy [Figure 1A]. Skull growth exceeding brain growth can occur in rickets, achondroplasia, and chronic hemolytic diseases and cause macrocephaly. The other mechanisms are 1) anatomical (neuronal and/or glial proliferation) as in neurocutaneous syndromes, overgrowth syndromes, Klinefelter syndrome and familial megalencephalia, 2) altered CSF dynamics (hydrocephalus) and 3) metabolic (lysosomal or demyelinating pathologies) such as mucolipidosis, mucopolysaccharidosis, sphingolipidosis, amino acid disorders, leukodystrophies, etc. Macrocephaly has also been associated with autistic spectrum disorders and chromosomal disorders. [3] These causes were ruled out in our case either clinically or on investigation. The clinical differentials initially did not include pontocerebellar hypoplasia because progressive macrocephaly is not known to be associated with it. There is only a single case report of a male child with pontocerebellar hypoplasia type 2 who had macrocephaly at birth but microcephaly at three months of age. [4] In conclusion, the association of progressive macrocephaly with pontocerebellar hypoplasia type 1 is unusual and similar reports may reinforce this association. References
Copyright 2009 - Indian Journal of Medical Sciences The following images related to this document are available:Photo images[ms09025f1.jpg] |
| |||||||||

{kind=link}