 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
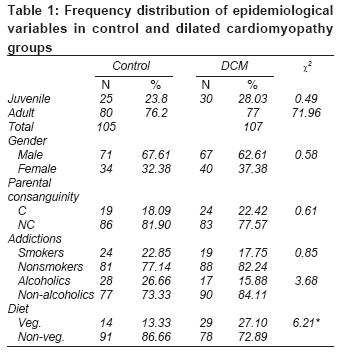
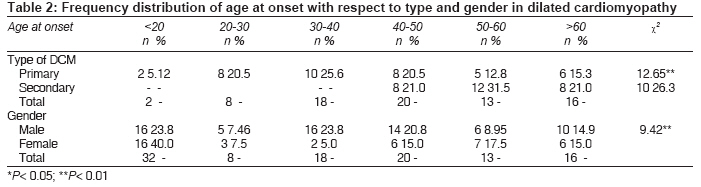
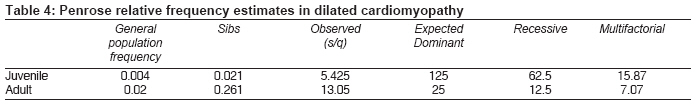
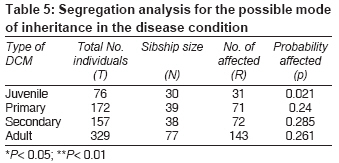
Indian Journal of Medical Sciences, Vol. 63, No. 7, July, 2009, pp. 288-296 Original Article Epidemiology and genetics of dilated cardiomyopathy in the Indian context Ushasree B, Shivani V, Venkateshwari A, Jain RK, Narsimhan C, Nallari Pratibha Department of Genetics, Osmania University, Hyderabad, Andhra Pradesh Code Number: ms090521 PMID: 19700909 DOI: 10.4103/0019-5359.55114 Abstract Background: Dilated cardiomyopathy (DCM) still remains to be a poorly understood and less analyzed group of cardiac-muscle disorders when compared to hypertrophic cardiomyopathy (HCM). Also, the vast clinical heterogeneity among the patients has rendered the small and isolated kindred studies less informative on the genetics and epidemiology of DCM.Aim of the study: The study aimed at understanding the epidemiology and genetics of DCMs in the Indian context. Materials and methods/ Statistical analysis: One hundred seven DCM patients and 105 healthy individuals were included in the study for epidemiological and genetic risk factor identification and to fit the possible mode of inheritance. Single's ascertainment methodology for segregation analysis and Penrose frequency estimates were followed to evaluate for the role of specific epidemiological factors in the disease etiology. Chi-square analysis was carried out to interpret the results statistically. Results and Conclusion: Our study suggests that epidemiological factors like gender, age at onset and vegetarian diet in conjunction with sarcomere gene mutations may play a role in the disease expression. Similarly, segregation analysis for the possible mode of inheritance showed a deviation from the autosomal dominant mode of inheritance, strengthening the underlying genetic heterogeneity of DCM. Keywords: Age at onset, dilated cardiomyopathy, gender, genetic heterogeneity Introduction Cardiomyopathy is the disease of the heart muscle, which causes deterioration of myocardial functioning. As per WHO criteria and American Heart Association/ European Society of Cardiology classification, cardiomyopathy is categorized as dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), obliterative cardiomyopathy (OCM) and arrhythmogenic right ventricular cardiomyopathy (ARVC). [1],[2],[3],[4] Of the above, DCM is the most prevalent but poorly understood disorder associated with sudden cardiac death. [5],[6],[7],[8] It is a heterogeneous group of cardiac-muscle disorders characterized by ventricular dilatation, impaired systolic function, reduced myocardial contractility and by inefficient pumping of the heart. [5] The cases were diagnosed as primary DCMs through coronary angiogram, wherein one can rule out the possibility of an underlying myocardial infarction/ coronary artery disease, valvular disease and/ or other systemic disorders. [9] However, hereditary and environmental factors, in combination with an altered immune response, appear to play a causative and pathogenic role in the etiopathology of dilated cardiomyopathy. The tissue damage caused by free radical generation and viral pathogenesis initiates a complex sequence of events, collectively known as inflammatory response. [9] To combat such responses, immunomodulators such as complement cascade system (C3,C4,C8) and alpha-1 anti-trypsin (AAT) were studied in the disease pathology for disease progression and its phenotypic expression. [10],[11],[12] These interactions contribute to the variable clinical expression of the disease among patients. While some individuals are asymptomatic, others may present with progressive refractory heart failure. DCM is a relatively common cause of heart failure, with a prevalence rate of 0.04% and incidence being 5 to 8 cases per 100,000 of population per year, increasing steadily. [8],[13],[14] Epidemiological studies carried out are limited to the western, European, Norwegian and Australian populations only. [15],[16] The incidence of the disease has been reported to vary from region to region and among ethnic groups. However, the true figure is likely to be higher as a consequence of under-reporting of mild asymptomatic cases, [17],[18] and the incidence varies according to the gender and ethnic background. The disease is reported to be more prevalent and aggressive in Blacks and in females, while a few other reports had shown a preponderance of males. [18],[19],[20] DCMs mostly follow autosomal dominant mode of inheritance with age-related penetrance, followed by autosomal recessive and, rarer, X-linked or mitochondrial modes of inheritance. [21] The inheritance pattern often depends on the specific gene involved in the disease causation, as well as on the degree of penetrance and expressivity of the gene concerned, which varies considerably among individuals of the same and/ or different ethnic groups and is age dependant, with a much higher incidence of the disease and severity of the condition at older ages. This indicates that an early diagnosis may alter the clinical course in familial dilated cardiomyopathy. Since the Indian population exhibits a great range of geographic and ethnic variation, along with an increased frequency of inbreeding, delineation of the mode of inheritance in conjunction with epidemiological variables like gender, age at onset, parental consanguinity, diet, etc., was examined in the Indian context. Materials and Methods Overall, 107 DCM patients in the age group of 3 months to 65 years, diagnosed according to WHO criteria (WHO 1996), referred to the cardiology units of CARE, KIMS and Niloufer Hospital for children, were included in the study. For comparative analysis, 105 healthy individuals with no known history of cardiac disorders in them or their family members were considered. Patients above 35 years of age had to undergo coronary angiography to rule out the possibility of ischemia/ coronary artery disease. The study was conducted in accordance with the declaration of Helsinki. The study protocol was approved by the concerned hospital and the university ethics committee, and subjects gave informed written consent before being enrolled for the study. Information on age, age at onset, duration of the disease, clinical symptoms, parental consanguinity, and history of smoking and alcohol consumption was also obtained from each subject. The cases were diagnosed as primary DCMs through coronary angiogram, wherein one can rule out the possibility of an underlying myocardial infarction/ coronary artery disease, valvular disease and/ or other systemic disorders. [19] Statistical analysis The data obtained on epidemiological variables were compiled by appropriate statistical/ genetic methodology such as Chi-square test of association for autosomal dominant mode of inheritance by segregation analysis following Single′s incomplete ascertainment method, and Penrose′s relative frequency estimates were calculated. Chi-square (χ2) test assuming autosomal dominant mode of inheritance was carried out [22] based on the following formula:
where ′A′ is the total number of affected individuals and ′N′ is the number of normal individuals. A significant deviation in the Chi-square value may suggest alternate modes of inheritance such as autosomal recessive/ X-linked or a multifactorial inheritance pattern. Segregation analysis was carried out to examine for the possible mode of inheritance, depending upon the sibship size and the proportion of affected individuals. Penrose′s method was adopted to calculate the relative frequencies based on the population frequency of the condition ′q′ and sibs′s′. The mode of inheritance was identified based on the comparison of the observed relative frequencies with those of the expected values, where the expected relative frequencies were calculated in sibs as 1/2q for an autosomal dominant trait, 1/4q for an autosomal recessive trait and 1/√q for a multifactorial trait. The observed frequencies are given by s/q. The calculated observed probability was examined for its close agreement to that of expected probability estimates of the dominant, recessive or multifactorial modes of inheritance. Single′s incomplete ascertainment method was followed to calculate the ′probability′ (′p′) value and variance component along with standard error to identify the modes of inheritance as suggested by Fischer. [23] p = R − N / T − N Variance = pq / T − N, where R = number of affected individuals in all sibships, T = total number of individuals in all sibships, N = number of sibships. The χ2 inference was made based on the estimates. Results and Discussion Epidemiological analysis [Table - 1] represents the frequency distribution of various epidemiological variables in the control and disease groups. Among the cohort of 107 patients, 28.03% constituted juvenile patients (6 months to 15 years), and 71.97% were adult DCM patients, in contrast to 23.8% of healthy juveniles and 76.2% healthy adult individuals categorized as control group. With respect to gender, 62.62% and 37.38% constituted male and female patients, respectively, with the male-female sex ratio being 2:1, unlike few studies which report female preponderance. [24] The preponderance of males could be explained on the basis of hormonal variations and genetic background, apart from differential life styles. Probably male hormones confer greater vulnerability to factors altering membrane integrity and permeability, [25],[26] as it is well established that estrogens are cardio-protective. [27] Parental consanguinity/ inbreeding was also documented in both control and disease groups [Table - 1]. Parental consanguinity was present in 24 (22.42%) of the 107 dilated cardiomyopathy patients, highlighting that the frequency of consanguinity is slightly higher in the disease group compared to that in the healthy control group (18.09%), which represents the local random population. This slightly higher parental consanguinity may be contributed partly by the juvenile and primary dilated cardiomyopathy cases in the present study. The above result is also partly strengthened by segregation analysis, wherein a subset of DCM seems to follow autosomal recessive mode of inheritance. Extensive clinical studies have identified factors like addictions to nicotine and alcohol as being contributory to cardiomyopathy. Nicotine and alcohol are known to increase the triglyceride levels in the blood and are associated with hypertension, arrhythmias and heart failure. In our study, smokers comprised 22.85% of the control group and 17.75% of DCM group in comparison to the nonsmokers, who comprised 77.14% and 82.24% of the two groups, respectively. However, the difference was not found to be significant statistically (χ2 0.85). Similarly, alcohol consumption was examined in both the control and disease groups, since it is the most common risk factor for DCM. In the present study, alcoholics comprised about 26.66% of the control group and 15.88% of DCM group in comparison to the non-alcoholics, who comprised 73.33% and 84.11% of the two groups, respectively, with no significant variation (χ2 3.68). Though it has been known that smoking and alcohol predispose to heart failure, [28],[29] our data showed no correlation of the above two epidemiological variables with the disease condition. The low percentage of smokers and alcoholics in DCM clearly reflects a subset of DCMs that are secondary to certain systemic disorders. With regard to diet, 86.66% of control group and 72.89% of disease group individuals were non-vegetarians, whereas only 13.33% of control group and 27.10% of DCM group comprised of vegetarians, with the difference being statistically significant (χ2 6.21), indicating that non-vegetarians may be at a higher risk for the disease condition, which can be correlated to high intake of polyunsaturated fatty acid. A non-vegetarian diet is a rich source of saturated fats but low in the nutritional supplements like protective antioxidant enzymes, polysaturated fats and fiber, which are essential to combat the oxidative stress. [30] This may lead to a general depletion in the antioxidant reserve, thereby making an individual more susceptible to the already existing disorder and exacerbating the disease condition. [Table - 2] reveals the frequency distribution of the age at onset with respect to type of dilated cardiomyopathy and gender, wherein the juvenile DCM patients′ age group varied from 3 months to 15 years. As many as 5.12% of individuals were <20 years of age, 20.5% were between 20 and 30 years and 25.6% belonged to the age group of 30-40 years, indicating an early age at onset for primary DCM, further supporting and strengthening the genetic basis of DCM. Similarly, the age at onset in secondary DCM showed that 31.5% of individuals belonged to 40-50 years age group, 21% were between 50 and 60 years of age and 26.3% belonged to the> 60 years age group, indicating the possible role of secondary risk factors like addictions to alcohol and smoking / other systemic and age-related systemic disorders like hypertension, diabetes mellitus in the disease pathophysiology. With regard to gender, 23.8% of the males belonged to 30-40 years age group, while 20.8% belonged to the 40-50 years age group, revealing that young adults and middle-aged men are prone to the disease, confirming the results of earlier studies carried out by the American Heart Association (1980). It has also been reported that men are at a higher risk of heart failure than women, [31] although the difference narrows with age, and that they may usually develop heart disease 10 to 15 years earlier than women. Social life style, like addictions to smoking and alcohol; heredity; and male sex hormones are known to play a role in the disease causation, affecting such characteristics as body fat distribution, etc., which are associated with an increased risk of heart disease, diabetes and stroke. With respect to age at onset in females, 40% and 17.5% of the female patients were below 20 years of age and above 50 years, respectively, indicating that women between 20 and 50 years of age have considerably low risk of developing the disease, which can be attributed to the role of cardio-protective hormones such as estrogen, which are known to have immunomodulatory and regulatory effects. [32],[33] The high preponderance of female patients below 20 years of age can be attributed to the genetic background with possible association of mutations of the sarcomeric genes; while the preponderance of females above the age of 50 years (17.5%) can be accounted by depletion of estrogen, a cardio-protective hormone, in the menopausal age group as suggested by many researchers. [34],[35],[36] This hypothesis is strengthened by recent studies, which revealed that estrogen therapy among older women with heart failure is associated with better prognosis. Genetic analysis [Table - 3] represents the segregation analysis to delineate the mode of inheritance in DCM, wherein juvenile DCM, primary DCM and secondary DCM cases were categorized on the basis of age and associated risk factors. The subtyping may help to delineate the extent of genetic component involved and the underlying genetic heterogeneity of the condition. Chi-square test was carried out to fit the autosomal dominant mode of inheritance, wherein the juvenile DCM (χ2 = 3.324; P< 0.05) showed no deviation from the assumed autosomal dominant mode of inheritance; while the primary (χ2 = 16.41; P< 0.05), secondary (χ2 = 12.95; P< 0.05), adult (comprising primary and secondary DCM cases combined) (χ2 = 28.73; P< 0.05) and pooled (χ2 = 31.28; P< 0.05) DCMs revealed a significant deviation from the autosomal dominant mode of inheritance, indicating alternate modes of inheritance. However, the number of juvenile cases that were reported to the hospital during the 4-year study was negligible. Hence a larger-sample study is recommended to substantiate the findings. [Table - 4] represents the Penrose relative frequency estimates to delineate and identify the genetic heterogeneity in DCM cases. The Penrose relative frequency estimate for juvenile DCM revealed the observed frequency (s/q) to be 5.425, while the expected frequency was found to be 15.87, which was estimated based on the general population frequency data. Similarly, with respect to adult DCM, the observed frequency was 13.05 and the expected frequency was found to be 12.5, revealing the mode of inheritance to be recessive in adult DCM cases, while it was multifactorial in JDCM. Thus the relative frequency estimates could partly delineate the underlying genetic heterogeneity of DCM. [Table - 5] presents the segregation frequencies to identify the mode of inheritance with respect to the subtypes of DCM. The analysis is based on single incomplete ascertainment method, wherein the sibship size and the total number of affected individuals were considered individually for each subgroup to obtain the respective probability estimates. The probability of being affected was observed to be 0.021 in juvenile DCM, 0.24 in primary DCM, 0.285 in secondary DCM and 0.265 in adult DCM cases. A deviation from autosomal recessive mode of inheritance in case of JDCM was observed on the basis of the estimates, while the adult DCM group seems to follow an autosomal recessive mode with incomplete penetrance, indicating that DCM is a heterogeneous-group disorder with the implication of different sarcomere and cytoskeletal genes/ variants in the etiology of the disease. Conclusion Dilated cardiomyopathy is a heterogeneous disorder with outcomes that depend on cause and age at presentation, as well as heart failure status. The study revealed male preponderance, with middle-aged men being affected preferably, while a majority of female patients belonged either to the juvenile group or to the postmenopausal age category. The Penrose relative frequency estimates revealed a possible multifactorial mode of inheritance in the case of juvenile DCM, and adult DCM cases seem to follow a recessive mode of inheritance. The test and the segregation analysis also revealed a deviation from the autosomal dominant mode of inheritance with incomplete penetrance and expressivity, supporting genetic heterogeneity of DCM. Acknowledgments The financial support from the Department of Biotechnology (DBT) and Indian Council of Medical Research (ICMR), New Delhi, India, for senior research fellowship (SRF) is gratefully acknowledged. References
Copyright 2009 - Indian Journal of Medical Sciences The following images related to this document are available:Photo images[ms09052t5.jpg] [ms09052t1.jpg] [ms09052t2.jpg] [ms09052t3.jpg] [ms09052t4.jpg] |
| |||||||||

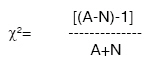
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}