 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
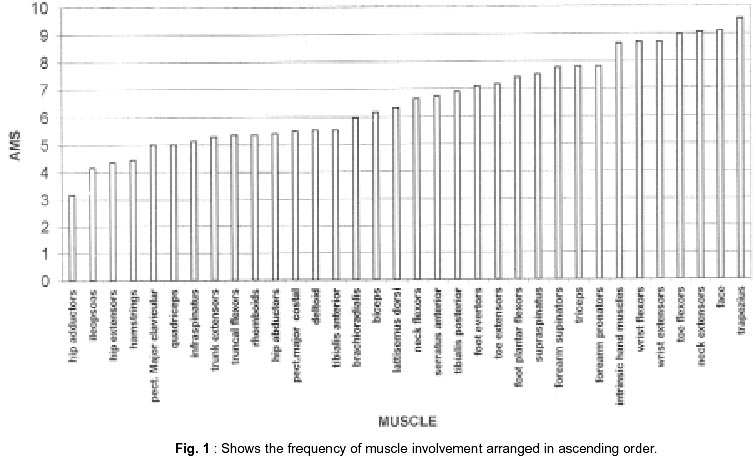
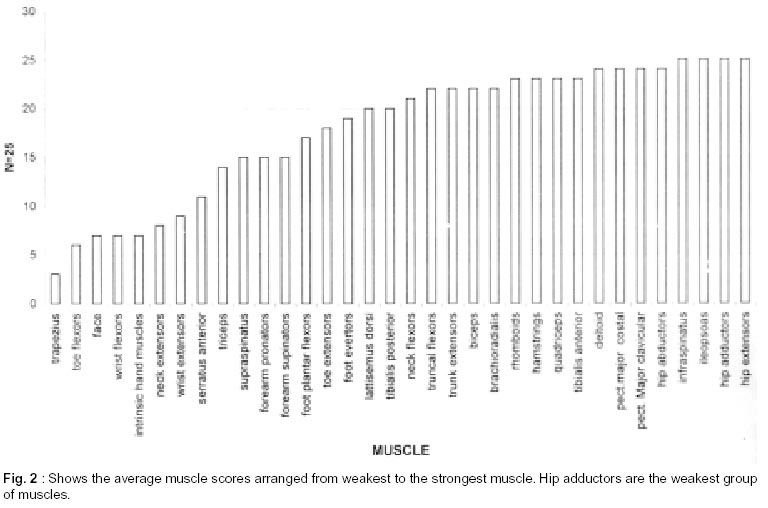
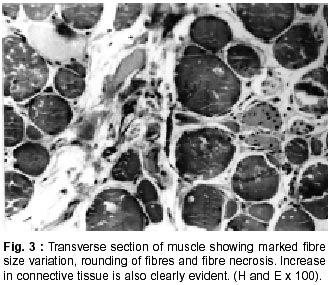
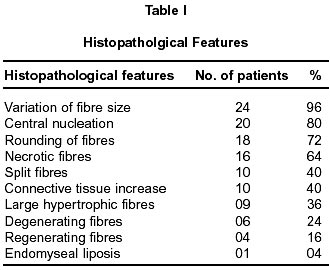
Neurology India, Vol. 50, No. 1, March, 2002, pp. 27-32 Sarcoglycanopathies : A Report of 25 Cases S.V. Khadilkar, R.K. Singh, S.M. Katrak Department
of Neurology, Sir JJ Group of Hospitals and Grant Medical College Mumbai - 400
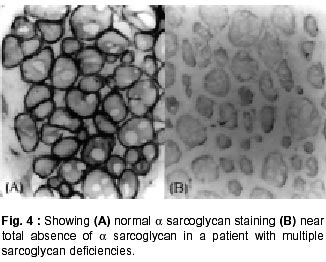
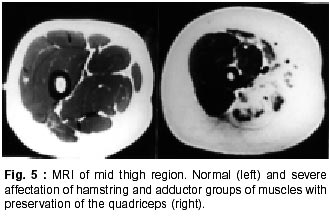
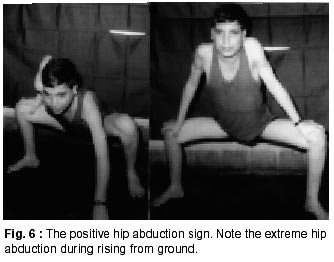
020, India. Accepted for publication : 12th September, 2001. Code Number: ni02008 Summary Twenty five patients with sarcoglycanopathies were studied prospectively. 21 of them had mild phenotype. Muscle involvement was more pronounced in adductor and flexor groups of muscles of the limbs, hip adductor muscles being the weakest. The selective and differential weakness between weak hip adductors and stronger hip abductors resulted in the hip abduction sign in 64% of cases. Distal muscle involvement in lower limbs was seen in 92% of cases, but was mild and late in the course of the disease. 44% patients had winging of scapulae. Immunocytochemistry showed multiple sarcoglycan deficiencies in 84% patients. Primary b and d sarcoglycanopathy was seen in the remaining 16% cases. Secondary dystrophin reduction was seen in 44% patients and correlated with b sarcoglycan deficiency but not with functional disability. Key words : Limb girdle muscular dystrophy, Phenotype, Sarcoglycan, Hip abduction sign. Introduction Limb girdle muscular dystrophy (LGMD) is a broad term encompassing a variety of muscular dystrophies presenting with limb girdle weakness. At the time of the original description by Walton and Nattrass1 in 1954, patients who could not be included in the better known dystrophies like the Duchenne muscular dystrophy and the fascio scapulo humeral muscular dystrophy were grouped as LGMD. Hence, the group consisted of many entities without clear characterization and the very existence of the term LGMD was doubted.2,3 However, with the inputs from immunocytochemistry and molecular biology, the scene has changed rapidly. Soon after the discovery of dystrophin, the dystrophin-associated glycoproteins (DAP) were purified. These proteins are expressed at the sarcolemma. The understanding of autosomal recessive LGMDs advanced with the elucidation of sarcoglycan transmembrane complex.4 Five sub units (a,b,g,d, and e) have been isolated and except for the epsilon sub unit, other four sub unit deficiencies have been shown to result in muscular dystrophies. The sarcoglycanopathy hypothesis states that the muscle degeneration in all the four sub units is by a common final pathway. Hence, the sarcoglycanopathies are considered as a complex.5 The aim of this study was to carry out prospective analysis of clinical features, biochemical, electrophysiological, histological and immunohistochemical characterization of patients with sarcoglycanopathies seen in Mumbai. Material and Methods The study was carried out at the neuromuscular clinic of a tertiary referral centre between 1996-2000. Patients were recruited as per clinical criteria for LGMD.6 Patients of either gender with chronic progressive limb girdle weakness, with onset in either pelvic or shoulder girdle or both, were included. Clinical exclusion criteria included congenital onset, ptosis or weakness of ocular muscles, onset in distal or facial muscles and rash compatible with dermatomyositis. All patients underwent detailed history and clinical neurological examination. Particular attention was given to the muscle strength and 32 pairs of muscles were tested in each patient. For the purpose of studying the pattern of muscular involvement, duration of illness was arbitrarily divided into less than or more than 5 years. Muscle strength was assessed using modified British Medical Council (MRC) grading.7 Muscle weaknesses of all groups of muscle were converted to 0-10 point system. The average muscle score (AMS) was derived from it. Thus, the weaker the muscle, the lower was the score.7 Hypertrophy and atrophy of the muscles was graded on inspection. Detailed pedigree charting was done. All available family members were examined. The investigations done included serum creatine kinase (CK), electromyography, nerve conduction studies and muscle biopsy examinations. Quantitative estimation of creatine kinase activity was done by dimension clinical chemistry system (UV enzymatic determination) using creatine kinase flex reagent cartridge. Electromyography and nerve conduction studies were done on Dantec system. Spontaneous activity, motor unit size, shape, interference pattern and amplitude and velocities of sensory and motor nerves were noted. Muscle biopsy was done in all patients. Biopsy specimens were snap frozen in chilled isopentane for frozen sections and later taken in formalin for paraffin embedding. Routine assessment of morphological changes was done on frozen and paraffin sections. Biopsy specimens were grouped into dystrophic, myopathic and mixed (neurogenic and myopathic) changes. Histochemical studies were done using ATPase and NADH -TR stains. Frozen sections of 6 microns were stained with dystrophin 1(C terminus-mouse monoclonal Dy8\6C5) and dystrophin 2 (Rod domainmouse monoclonal Dy4\6D3) and a (Ad1\20A6), b (Bsarc\5B1), g (35 DAG\21B5) and d (Dsarc 3\12C1) sarcoglycan antibodies (Novocastra labs), using peroxidase method and standard controls. MRI of thigh muscles was performed in only 2 patients, using 1.0 tesla super conducting imaging system body coils. Results Preliminary Data : Seventy six patients fulfilled the clinical inclusion criteria and 54 of them underwent full investigations including immunocytochemistry. Out of these 54 patients, 25 patients were detected to have sarcoglycanopathies and they formed the basis of this study. There were 13 female and 12 male patients. 13 patients had positive family history, with autosomal recessive mode of inheritance and the remaining 12 were sporadic (3 males and 9 females). Mean age of the patients was 25.84 years (range 8-42 years) and mean age of onset of illness was 15 years (range 5-37 years). Mean duration of illness was 7.6 years (range 2-22 years). Nine patients presented within 5 years and 16 patients after 5 years. Twenty one patients were ambulant when examined and the remaining 4 were wheelchair bound. Observations on Clinical Phenotype : Weakness initially involved the pelvic girdle and later the shoulder girdle. Selectivity of muscle weakness was more conspicuous in mild to moderately weak patients. Fig. 1 shows the frequency and Fig. 2 the severity of muscle weakness. Hip adductors were the weakest (AMS: 3.16) group of muscles. 68% patients had disproportionate hip adductor weakness compared to abductors of hip. Hamstrings were weaker than the quadriceps (AMS 4.4, 5 respectively). In the upper limbs, biceps, deltoids, pectoralis major, rhomboids and infraspinatus were the more affected muscles. Even in those patients who presented early in the illness, severe weakness of hip adductors, biceps and hamstrings was evident (Fig. 3). Tibialis anterior was the most frequently involved distal muscle, but to a lesser degree compared to its proximal counterparts (AMS 5.5). 4 patients (16%) had predominant gastrocnemius weakness. 11 patients (44%) had scapular winging. Trunc flexors and paraspinals were as weak as pelvic girdle muscles. Mild facial weakness was noted in 7 patients (28%). None had pharyngeal or lingual weakness. Atrophy was noted in thigh adductors (44%), hamstrings (20%) and calves in 20% of cases. Distal upper limb wasting was present in 4 cases. Apparent prominence of calves due to wasting of the proximal muscles was observed in 40% patients. Gait analysis revealed that most of the patients (96%) waddled only minimally. Severe symptomatic cardiomyopathy was present in one male, aged 14 years. 28 Laboratory Data : Serum CPK activity ranged from 208-15,571 IU, being higher with earlier presentation. Electrophysiology was abnormal in all the patients and showed short duration, low amplitude polyphasics with full recruitment, suggestive of primary muscle disease. 58.8% muscles showed spontaneous activity. In 8 patients, with disease duration of less than 5 years, the adductor and hamstrings muscles were electrophysiologically more severely affected than quadriceps. In one female, only adductors showed electrophysiological abnormalities. Histopathological evaluation was consistent with dystrophy in all patients (Table I, Fig. 3). Results of immunohistochemical study showed multiple sarcoglycan deficiencies in 84 % patients (Fig. 4a and b). Amongst them, all sub units were deficient in 24%, three in 24% and two in the remaining 36% patients. Primary b and g sarcoglycan deficiency was noted in 12% and 4% patients respectively. Secondary partial dystrophin deficiency was seen in 44% patients. Overall, sarcoglycan reduction was most common (64%) and it was deficient in all except one of the patients having dystrophin deficiency. MRI study of thighs and calves was done in two patients. The mid-thigh section showed wasted adductor compartment and hamstrings with preservation of vasti (Fig. 5). Discussion In the present study, male to female ratio was almost equal, which is consistent with the autosomal recessive mode of inheritance of sarcoglycanopathies. Autosomal recessive transmission was established in 52% of our patients. Remaining 48% appeared sporadic. Complete investigations of family members may have yielded more familial cases. However, this was not feasible in this study. Twenty one patients had mild phenotype. The severe Duchenne like phenotype was seen only in four patients. The authors believe this to be a reflection of the referral pattern as the percentage of pediatric cases referred to the clinic was small. Differential and selective muscle involvement is an important feature of muscular dystrophies and highlights the phenotype. In the patients presented here, adductor and flexor groups of muscles were generally more severely involved than abductors and extensors. Hip girdle muscles were the weakest. A striking feature was early and severe involvement of hip adductors compared to the abductors, which remained relatively spared till late in the disease. Disproportionate hip adductor weakness was evident even in the first or second year of symptomatic stage of illness. In 64% of patients, the combination of weak hip adductors and strong abductors resulted in splaying out of thighs while rising from ground, the 'hip abduction sign'8 (Fig. 6). The hip girdle weakness and splaying of thighs is noticed early by Indian patients, as they squat to defecate. Our patients had minimal waddling, indicating that hip abductors were relatively strong. Adductor weakness has been previously noted in childhood onset g sarcoglycanopathies, 9,10 in adults11 and in a sarcoglycanopathies. 12,13 We wish to stress that the early and severe weakness of hip adductor muscles with relative preservation of the abductors is a characteristic feature of this phenotype. Another feature was prominent weakness of hamstring muscles, with comparatively stronger quadriceps. This is in keeping with the general pattern of flexors being weaker than extensors and is in concurrence with other studies.14 The relative sparing of quadriceps should be taken into consideration when choosing biopsy site for routine histology. Selectivity of muscle weakness was evident even early in the course of the disease. Distal muscle weakness, particularly tibialis anterior, was very common. It was involved nearly as frequently as proximal hip girdle muscles but in later stages and to a milder degree. Predominant gastrocnemius weakness was noted in only four patients. Although recently noted in dysferlinopathy,15 this specific involvement has not been stressed in sarcoglycanopathies. Scapular winging was noted in 44% patients. The presence of scapular winging and distal muscle weakness may simulate other scapuloperoneal syndromes. However the selective and severe proximal hip girdle involvement distinguishes these patients. Severe cardiomyopathy was encountered in one of our patients, a 14 year old male with severe clinical phenotype. He showed d and b -SG deficiencies. Mutation of either b , g or d sarcoglycan can lead to cardiomyopathy. However, in humans, d sarcoglycan gene mutation leads to familial or sporadic cardiomyopathy without involvement of the skeletal muscle. Hence, in him, b -SG might have been the primary deficiency.16 The CPK levels correlated with stage of the illness i.e. levels decreased with advancing illness. Electrophysiology was consistent with the myopathic process. In early cases it was noted that adductors and hamstring muscles tended to show severe changes, in tandem with the phenotypic and MRI findings. The most frequent immunostaining pattern was of multiple sarcoglycan deficiencies in various combinations. b -SG was the most frequently deficient fraction. This finding is logical, as b. SG along with d SG forms the core of the SG-complex. It has also been reported that b , d and a -SG are closely associated with each other and hence are likely to be deficient together.17 However we did not detect this pattern of deficiency in our patients. Population based studies have shown that a -SG deficiency is the commonest.18,19 Our patients had mainly b -SG deficiency. This may be partly explained by the fact that patients in our study had mild phenotype whereas a sarcoglycanopathy presents early and has a severe phenotype like Duchenne muscular dystrophy.14 Secondary dystrophin deficiency is well known in patients with sarcoglycan deficiency and is seen more often with g -SG deficiency,18 as dystrophin is thought to anchor g -SG in SG-complex. However in 11 of our patients with relative dystrophin deficiency, 10 had b -SG deficiency, thus signifying the complexity of interactions between dystrophin and SG-complex. Secondary dystrophin reduction has also been implicated in the process of muscle degeneration. However we did not find any correlation between histological severity of the dystrophic process with the reduction in dystrophin levels. Conclusion In this small series, a distinct phenotypic pattern of muscular weakness emerges. Characteristic feature of the phenotype is the early and severe involvement of the hip adductors, hamstrings and biceps muscles. The weakness of hip adductors with the relative preservation of abductor muscles results in the characteristic hip abduction sign, a clinical pointer to the diagnosis of sarcoglycanopathies. Severe Duchenne-like phenotype was uncommon. Immuno- staining showed multiple sarcoglycan deficiencies. A larger series may shed more light on clinical features of individual sub unit deficiencies. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02008t1.jpg] [ni02008f6.jpg] [ni02008f5.jpg] [ni02008f4.jpg] [ni02008f2.jpg] [ni02008f1.jpg] [ni02008f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}