 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 50, No. 1, March, 2002, pp. 45-52 Hypertrophic Pachymeningitis : Varied Manifestations of a Single Disease Entity S. Prabhakar, R. Bhatia, V. Lal, Paramjeet Singh* Departments of Neurology
and Radiodiagnosis*, Postgraduate Institute of Medical Educaion and Research,
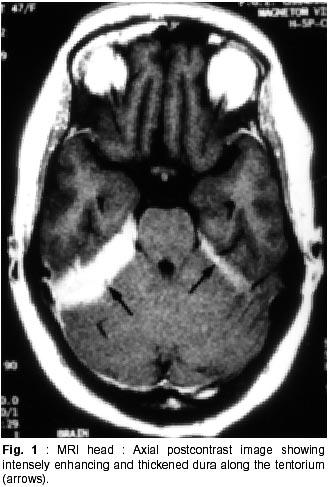
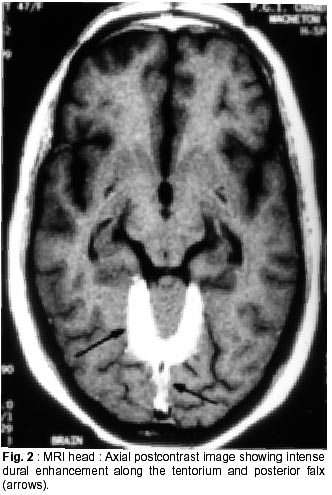
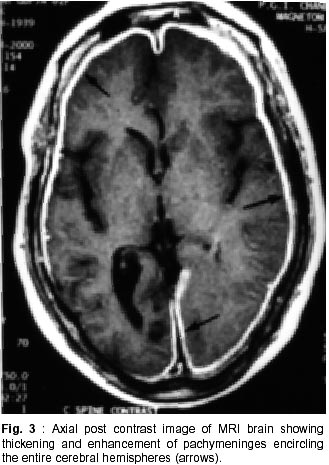
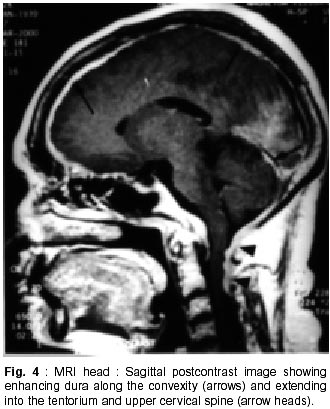
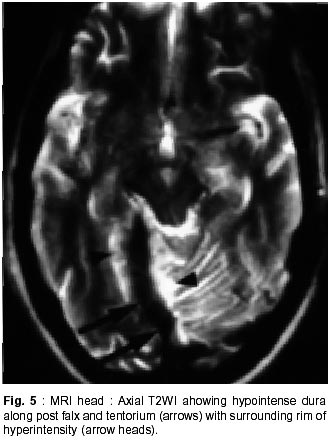
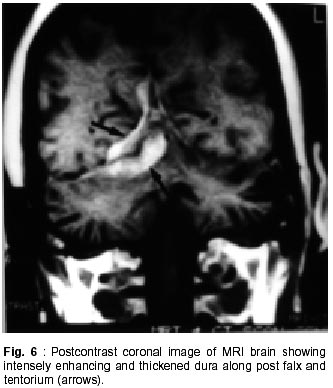
Chandigarh - 160 012, India. Accepted for publication : 18th December, 2000. Code Number: ni02012 Summary Hypertrophic pachymeningitis is a unique clinical entity characterised by fibrosis and thickening of the duramater with resulting neurological dysfunction. Three cases of this entity are described. Presenting features were headaches and cranial neuropathies in two patients and predominantly cerebellar dysfunction in the third. One of the patients also had evidence of spinal involvement. Lower cranial nerves were chiefly involved in two patients whereas optic nerve was the predominantly affected nerve in one. Except for the presence of rheumatoid arthritis in one of the patients, we could not document clinical or biochemical evidence of any predisposing infective, inflammatory or infiltrative condition in the other two. All three patients had characteristic changes on imaging suggestive of thickened and enhancing duramater. Although variable steroid responsiveness was seen in all the three patients, tendency towards steroid dependance was evident. The clinical presentations, causes, radiological features, management options and differential diagnosis of this unique clinical syndrome have been discussed. Key words : Pachymeningitis, Cranial neuropathies. Introduction Hypertrophic pachymeningitis is an uncommon disorder characterized by thickening and fibrosis of the duramater. A variety of inflammatory and infectious conditions can result in this condition. The condition is labelled as 'idiopathic hypertrophic pachymeningitis' in the absence of any definite inciting factor. Common clinical features include headaches, multiple cranial nerve palsies and cerebellar dysfunction ocurring alone or in combination. Three cases of this unique clinical entity are described and clinical features, radiological findings and therapeutic options are discussed. Case Reports Case 1 : BB, 47 year female presented with history of recurrent cranial neuropathies. Her symptoms started in December'97, when she developed tinnitus and bilateral hearing impairment without any other neurological deficit. Symptoms improved on some treatment, details of which were not available. In May'98 she had increasing temporo-occipital headaches, accompanied with right sided hearing deficit. In August'98 she had worsening headaches and developed bulbar weakness in the form of dysphagia, nasal regurgitation of fluids, dysphonia and dysarthria. Within few days she developed diplopia with a left 6th nerve palsy. Evaluation did not reveal any biochemical or CSF abnormality and cranial imaging was reported as normal. She received steroid therapy and showed excellent improvement in her symptoms. In February'99 she was admitted with increasing headache, dysphagia, hoarseness of voice and gait disturbance. There was no history of visual blurring, motor weakness or sensory deficit. There were no other systemic complaints. She had history of recurrent migrainous headaches for the last 20 years and was diagnosed to have hypertension six months back. Examination revealed cushinoid facies and a normal chest, CVS and abdomen examination. Neurological examination revealed a gaze evoked nystagmus, broken pursuits and dysmetric saccades. She had 6th, 9th and 10th nerve paresis, and gait ataxia with minimal limb ataxia on the right side. Investigations revealed an ESR of 52 mm and negative ANA, ANCA and RA factor. Serum ACE levels were normal. Results of Mantoux and pathergy test did not reveal any abnormality. CSF examination showed absence of pleocytosis, a protein level of 30 mg% and normal sugar. Gram's stain, fungal smear and antigen detection and staining for AFB were negative. Cultures for fungus and bacteria showed absence of any growth. Serum and CSF VDRL tests were negative. CSF ADA levels were 5U/L. X-ray chest, paranasal sinuses and high resolution CT of temporal bone were normal. MRI head (plain and contrast) revealed diffuse thickening and enhancement of pachymeninges along both sides of tentorium, posteroinferior falx, and along petrous dura with greater involvement on the right side (Fig. 1, 2). The patient did not agree for biopsy. Considering the history and results of other investigations, a tentative diagnosis of possible 'idiopathic hypertrophic pachymeningitis' was made and she was started on steroid therapy. Within days she showed a marked improvement in her clinical symptoms. She had relapse of symptoms on stopping steroids on two occasions with no clinical evidence of any new symptomatology. Presently, she continues on low dose alternate day steroid therapy with complete remission of symptoms. Case 2 : KG, 55 years female, was a known case of rheumatoid arthritis for the last 15 years, for which she had received NSAIDs, steroids and gold therapy. The symptoms were quiescent for last two years on antiinflammatory drugs. She developed two episodes of hearing loss from left ear in the last 10 years which had completely recovered. Since three years, she was having episodic headaches, chiefly in the right frontoparietal region without associated nausea or vomiting. Three years back, she developed acute onset visual loss from the left eye with mild improvement. Two years back, she developed reduced vision from her right eye, for which she received methylprednisolone therapy and showed significant improvement in visual acquity. Four months prior to admission she developed worsening of vision from her right eye. She received another course of methylprednisolone therapy with moderate improvement in her symptoms. During this period she was having increasing frequency and intensity of headaches and intermittent low grade fever for which she was started on antitubercular therapy. Three weeks before admission she developed dysarthria, dysphonia, dysphagia and pain in the right upper limb accompanied by weakness and heaviness. There were no bladder or bowel disturbances, sensory impairment or mental status abnormalities. There was no increase in symptoms of arthritis or evidence of rash or skin changes. There was no history of nasal discharge, hemoptysis, or weight loss. She was admitted under neurology services for evaluation. General physical examination revealed joint deformity in both hands and mild anemia. Neurological examination revealed bilateral optic pallor, left 6th and bilateral 8th nerve palsy. There was mild proximal weakness in all four limbs with attenuation of reflexes in right upper limb. Investigations revealed high ESR, and normal hemogram, renal and liver function tests. Rheumatoid factor was positive, although results of ANA, LE cell and ANCA were negative. CSF analysis showed a lymphocytic pleocytosis with normal sugar and protein levels. Gram's stain, fungal smear and stains for AFB and malignant cells were negative. Culture for micro-organisms was sterile. ADA levels were 4 U/L. Nerve conduction studies revealed absence of F responses. MRI head (plain and contrast) revealed thickening and enhancement of pachymeninges surrounding the entire brain, extending into the left tentorium and around the cerebellum, upto the upper cervical spine. (Fig. 3, 4). MRI of the dorsolumbar spine was normal. The patient was started on oral steroid therapy. There was improvement in bulbar symptoms and limb weakness, although improvement in visual acquity was minimal. She continues on low dose steroid therapy, with disease being stable till the last follow-up. Case 3 : BS, 54 years male, presented with features of progressive limb and gait ataxia, dysarthria and intention tremor of two years duration. Two months before admission, he developed difficulty in swallowing and hoarseness of voice. He had intermittent non-throbbing occipito-nuchal headaches, without any associated nausea or vomiting. There were no accompanying motor symptoms, sensory deficit or bladder disturbances. There was no history of fever, loss of weight, appetite or other features of active vasculitis or connective tissue disease. There was no past history of diabetes or tuberculosis. He was a known case of hypertension and coronary artery disease on therapy. Examination revealed mild pedal edema. Neurological examination revealed right IX, X cranial nerve paresis and bilateral cerebellar signs. Investigations revealed an ESR of 56 mm, normal hemogram, renal and liver function tests and a PSA level of 6.6 ng/ml. ANA, LE cell, ANCA,and RF tests were negative. Serum ACE levels were not elevated. CSF examination revealed absence of pleocytosis, protein level of 35 mg% and normal sugar. Reports of India ink examination, Gram's stain, AFB and cultures for fungi, bacteria and acid-fast bacilli were negative. No evidence of malignant cells was found on two occasions. X-ray chest was normal and abdominal ultrasound revealed mild prostatomegaly. MRI scan of the head revealed marked dural thickening along tentorium and posterior falx. It was hypointense on both T1 and T2WI with a surrounding rim of hyperintensity on T2WI (Fig. 5). Contrast enhancement was prominent (Fig. 6). In addition to these findings few hyperintensities were seen in superior vermis and inferior cerebellar peduncle. The patient was initially started on azathioprine therapy. Steroids were added later because of minimal improvement with azathioprine. There was gradual improvement in his clinical condition. Presently, he continues on alternate day prednisolone and azathioprine therapy. He has shown slow improvement in symptoms. There have been no fresh deficits or new clinical symptoms to suggest an alternate diagnosis. Discussion Hypertrophic pachymeningitis is a fibrosing inflammatory process that thickens the dura mater.1 Initial description of this process was made by Gowers, who divided it into external and internal forms, based upon various inciting factors.2 However, features matching current descriptions of this entity were possibly described by Charcot and later by Nafziger et al.3 Broadly, the condition may be described as 'primary' or 'idiopathic hypertrophic pachymeningitis' where no identifiable cause is found and 'secondary' where identifiable causes co-exist, although their definite relationship to the development of this condition may be debatable.1 Idiopathic hypertrophic pachymeningitis is, thus, a diagnosis of exclusion.4 Exact etiopathogenesis of this entity is still unknown. It might be an autoimmune phenomenon or occur as a direct result of infectious or infiltrative pathology.2,4 Common end result is encasement of brain tissue by thickened and fibrous meninges with resultant clinical symptomatology. The disease may be focal or multicentric in presentation. Clinical symptomatology in majority of patients is similar, although no single feature is specific for the disease. Chief symptoms are headache, cranial neuropathies and cerebellar dysfunction, occuring either alone or in combination.2,5,6 Spinal pachymeningitis with manifestations of radiculopathy, myelopathy or a combination of the two has also been observed. It either occurs alone or as a craniospinal form.7,8 Headache is a universal symptom, which can be focal or diffuse and at times may be the only symptom for years before other symptoms manifest.9 Two of our patients had prominent headaches especially during increase in the severity of other symptoms. Long history of migrainous headaches was observed in one of the patients. Since we could not document raised ICP or hydrocephalus in any of the cases, the headache possibly denotes dural inflammation.5 Cranial neuropathies were present in all the cases. Lower cranial nerve involvement, especially VIII and X, was most commonly observed. Almost any cranial nerve except I can be involved.2 Two patterns of cranial nerve involvement have been described, based on the site of dural inflammation : cavernous sinus to superior orbital fissure involvement and falcotentorial to posterior fossa dural involvement.2,5,10,11 Anterior involvement presents with paresis of II-VI cranial nerves, and can simulate a Tolosa-Hunt syndrome, thereby suggesting a possible relationship between the two.10,11 Posteriorly, cranial nerves V to XII may be involved, with involvement of VIII nerve being the commonest.2 Dense fibrous encasement and ischemic damage by hypertrophic tissue is the likely explanation for the cranial nerve deficits. Recurrent visual loss due to optic neuropathy was observed in one of the patients. Overall, occurrence of this phenomenon is uncommon.4,5,12,13 Meningeal thickening, raised ICP and recently described central retinal vein occlusion,14 have been proposed as possible mechanisms. Presentation with cerebellar dysfunction has been reported previously,2 although frequency of occurrence is low. Diffuse ischemia, venous sinus congestion and mass effect of thickened tentorium have been labelled as plausible explanations. Hyperintense signals in middle cerebellar peduncle and vermis were documented in one of our patients, which could suggest an ischemic damage to the underlying tissue. Uncommon clinical presentations include encephalitis,15 hypopituitarism, 16 diabetes inspidus,5 sinus thrombosis17 and hydrocephalus.18 Spinal pachymeningitis commonly involves the cervico-dorsal dura and presents with features of radiculopathy and/or myelopathy,7,8 either at single or multiple levels. One of our patients had weakness of right arm, attenuated reflexes, absent 'F' waves responses and enhancing dura upto C3 level on imaging, possibly suggesting associated spinal involvement. Investigative work up of this condition aims at excluding all possible causes of infectious, noninfectious and malignant etiologies. This includes biochemical work up, imaging studies and detailed evaluation of a particular disease process when suspected. This would include systemic evaluation for granulomatous diseases such as sarcoid or tuberculosis, vasculitis, connective tissue disease, and work up for a metastatic or infiltrative malignancy. Elevation of ESR is common. Tests like ACE levels, vasculitic profile, Mantoux test and cultures for bacteria and fungi should be done to rule out specific disease states. Except for the presence of positive rheumatoid factor in one patient with co-existent rheumatoid arthritis (RA), we could not document a specific abnormality in any of the patients. CSF is an important investigation, although studies are inconclusive. There is usually a lymphocytic pleocytosis with variable increase in cell counts.1,2,5 Protein levels are moderately elevated. However, CSF study may be entirely normal in many patients. One of our patients showed lymphocytic pleocytosis but levels of protein and sugar were normal in all. No abnormal cells were identified on flow cytometry or staining. No micro-organism was identified in any of the samples by smear or culture. Imaging is an important investigation to identify a meningeal based pathology and exclude mass lesions in the brainstem or skull base. All three patients showed prominent pachymeningeal changes on imaging. Thickening of dura mater was observed in all three. Tentorial and posterior falx involvement was seen in two of the patients. One of them showed diffuse thickening of dura encasing the entire brain. MRI showed hypo-isotense dura on T1 and T2WI and marked enhancement after contrast administration. The area of involvement correlated with the clinical picture in all the three patients. Features observed were quite similar to that mentioned in the literature.4,5,19,21 Non-enhanced CT scans show thickened, hyperdense dura, typically along the tentorium, tentorial ridge, falx and prepontine brain stem with marked enhancement on contrast administration. The thickened dura on MR images appears isointense to hypotense on both T1 and T2W images and usually shows a uniform dense enhancement of the thickened membranes.4,19 The thickening is better appreciated on coronal and sagittal images. Thin rim of hyperintensity may be present around the hypointense dura in some patients2,4,19 as appreciated in one of our cases (Fig. 5). Two patterns of enhancement, namely linear and nodular have been described with the former showing better and sustained therapeutic response, possibly related to less fibrosis and more vascularity. The changes on imaging are probably related to the presence of dense fibrous tissue, with decrease in interstitial space and paucity of imageable free water, accounting for the hypointensity on both T1 and T2W images. Presence of associated leptomeningeal enhancement or parenchymal abnormalities should suggest an alternate diagnosis.4 A dural biopsy is considered important for differentiating idiopathic from secondary forms of pachymeningitis.1,2,6,18 Since many associations and secondary causes for pachymeningeal thickening and enhancement exist, pathological examination of meninges is essential. Pathological features include thickening, fibrosis and presence of inflammatory cells including plasma cells and lymphocytes. Presence of granulomas or vasculitis aids in establishing a specific etiology. None of our patients agreed for a dural biopsy. However, clinical symptomatology, imaging characteristics, absence of abnormal laboratory and CSF studies, long course of the disease and responsiveness to steroid therapy point towards an idiopathic variety in two of our cases. One of the patients had concomitant rheumatoid arthritis as a possible etiological factor, although the relationship is not entirely proven. There is a known association between clinical RA22 and presence of RA factor alone.23 However, a meningeal biopsy may not reveal any evidence of vasculitis or rheumatoid nodules,22 further adding speculation to the relationship between the two. Differential diagnosis of this condition includes all pathological conditions that produce pachymeningeal thickening and have similar clinical and imaging features. As mentioned previously, the diagnosis of idiopathic form is one of exclusion. Tuberculosis should be considered in the differential diagnosis of any dural based pathology. Although commonly presenting as leptomeningitis and tuberculomas, pachymeningitis has also been reported in the literature24-26 and considered a distinct entity. Clinical features and imaging characteristics may be similar to the idiopathic form and evidence of tuberculosis may be found elsewhere in the body.24 However, biopsy may not always reveal granulomas or a positive AFB stain, and response to therapy has been taken as a criteria to label imaging characteristics of pachymeningitis as tubercular in origin.24 Syphilitic pachymeningitis,27 can be excluded by serological tests and cultures for Treponema pallidum on biopsy samples.2 None of our patients had a positive serology for syphilis. Neurosarcoidosis causes non-caseating granulomas with predilection for facial nerve, usually involved bilaterally.28 Other important presentations of neurosarcoidosis include basal meningitis, chronic aseptic meningitis and hypophysial dysfunction. However, isolated neurosarcoid occurs in only 2.5% of cases28,29 and sarcoid pachymeningitis is still uncommon. Abnormalities on chest X-ray, elevated serum and CSF ACE levels and a better and sustained response to steroids help in recognizing this entity. None of these features were present in our patients. Features of Wegener's granulomatosis were not present in any of the patients reported here. It usually presents with sinus and nasopharyngeal pathology although association with pachymeningitis has been observed.30,31 Cranial neuropathies occur mostly due to contiguous involvement from sinuses. Associated mononeuritis multiplex has been reported in upto 28% of the patients. The progression of this disease is more rapid and potentially fatal as compared to hypertrophic pachymeningitis of uncertain etiology. Evidence of necrotising vasculitis on biopsy, and positive ANCA can aid in establishing the diagnosis. Tolosa-Hunt syndrome, characterized by painful ophthalmoplegia, is an important differential diagnosis.37 It has been postulated that cranial pachymeningitis and Tolosa-Hunt syndrome represent facets of a common dural inflammatory reaction to yet unknown inciting factors. None of our patients had clinical and imaging characteristics of this entity. Intracranial hypotension is an important cause of meningeal enhancement.33 It presents with postural headaches, occasionally abducens palsy and low CSF pressures. The condition occurs spontaneously or follows craniotomy, shunt placement, lumbar puncture, head trauma or dural tears. The intense dural enhancement is possibly related to compensatory increase in venous blood secondary to reduced CSF volume and pressure. Presence of normal CSF pressure, pattern of presentation and responsiveness to steroids are features against this diagnosis in our cases. Neoplastic meningitis needs to be excluded in any patient with headache, cranial nerve palsies and meningeal enhancement. Lymphoma, leukemia, adenocarcinoma and melanoma can involve the meninges diffusely.34 However, negative cytology in CSF and clinical course of the patients with remission and relapses argues against this possibility in the present observation. Hypertrophic pachymeningitis might also be related to a group of disorders with fibrosclerotic tendency2 e.g. sclerosing cholangitis, episcleritis etc. None of our patients had evidence of a generalised fibrotic process. En-plaque meningiomas can infiltrate along the dura of the base of skull, into the frontal, temporal, orbital and contralateral sphenoid regions, resulting in multiple cranial nerve palsies and an orbital syndrome. All our patients were treated with steroid therapy. Clinical improvement was excellent in one and moderate in two patients. One of the patients had relapses on steroid withdrawl and presently continues on low dose alternate day therapy with complete remission of symptoms. Another patient received azathioprine to begin with. However, clinical improvement was poor and he was later started on steroids with improvement in symptoms. Presently, he continues to be on combined low dose steroids and azathioprine therapy. The lady with associated RA received ATT empirically without any improvement, which was withdrawn after admission to our institute. She had received methyl prednisolone twice in between with improvement of symptoms. Although, her clinical improvement has been moderate on low dose steroid therapy, no worsening of symptomatology has occurred. Steroid therapy has been considered the mainstay of therapy in this disease. However, the disease progression might continue despite institution of steroids and many patients may eventually become steroid dependent, as observed in our patients.2,5 Pulse therapy with methyl prednisolone has been advocated to reduce side effects and increase efficacy. Immunomodulating agents like azathioprine and cyclophosphamide have been tried, considering an autoimmune basis, with reports of improvement, although the efficacy has not been proven. Combined therapy with steroids and azathioprine has been used with improvement.35 Long term improvement with lymphocytopharesis has been shown in one patient recently.36 Considering occult tuberculosis, empirical use of antitubercular therapy has been advocated37 in patients with pachymeningeal enhancement on MRI, inspite of normal histology and absence of peripheral tuberculosis with reports of marked improvement in symptoms.19 One of the patients discussed here, received 4 months of ATT without any benefit. Although this part of the world has high incidence of tuberculosis, empirical use of ATT may not be justified. Surgery has both therapeutic and diagnostic benefits.5 Decompression has been used for spinal and orbital lesions.2,5,38 Radiation therapy has been used in the past without any proven benefits.2 Thus, hypertrophic pachymeningitis is an important cause of recurrent cranial neuropathies and headaches. Many infectious, inflammatory, non-infectious and malignant conditions can produce a similar picture, although the patterns of imaging characteristics and laboratory investigations help in differentiating between them. Idiopathic variety is usually responsive to steroids and specific therapy for secondary causes needs to be instituted. Aim of the therapy is to prevent permanent damage to neural structures. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02012f5.jpg] [ni02012f2.jpg] [ni02012f6.jpg] [ni02012f4.jpg] [ni02012f3.jpg] [ni02012f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}