 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 50, No. 1, March, 2002, pp. 53-59 Idiopathic Hypertrophic Cranial Pachymeningitis P.N. Sylaja, P.J. Cherian, C.K. Das,* V.V. Radhakrishnan,** K. Radhakrishnan Departments of
Neurology, Radiology* and Pathology**, Sree Chitra Tirunal Institute for Medical
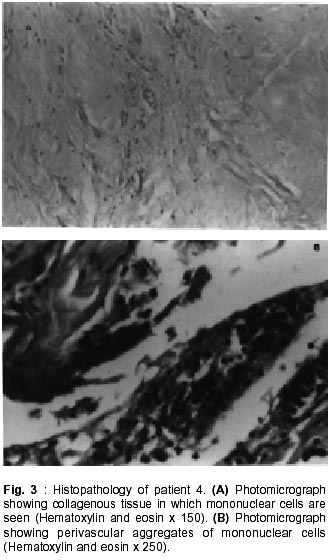
Sciences and Technology, Trivandrum, Kerala, India. Accepted for publication : 22nd November, 2000. Code Number: ni02013 Summary Idiopathic hypertrophic cranial pachymeningitis is a rare form of fibrosing chronic inflammatory process of unknown etiology, which causes thickening of the intracranial dura mater. We present four patients with hypertrophic cranial pachymeningitis who presented with chronic headache and cranial nerve palsies. The diagnosis of idiopathic hypertrophic cranial pachymeningitis was based on neuroimaging findings of thickened enhancing dura, exclusion of known causes and histopathologic findings compatible with nonspecific inflammation in the meningeal biopsies. Corticosteroid therapy was effective in all cases in inducing a complete or partial remission of the neurologic symptoms and signs. We describe the clinical, radiological and pathological features of idiopathic hypertrophic cranial pachymeningitis and discuss the relationship of this entity with other inflammatory fibrosclerotic disorders to explain the pathogenesis. A high index of suspicion, prompt confirmation of the diagnosis by meningeal biopsy, and early institution and long-term maintenance of steroid therapy may help to prevent irreversible neurologic sequelae, especially blindness. Key words : Cranial nerve palsy, Headache, Hypertrophic cranial pachymeningitis, Meningitis, Pachymeningitis. Introduction Hypertrophic cranial pachymeningitis is a rare chronic fibrosing inflammatory disease characterized by marked diffuse thickening of the cranial dura mater that causes progressive neurological deficits.9,13 Chronic headache and cranial neuropathy are the common clinical manifestations of patients with hypertrophic cranial pachymeningitis. Numerous clinico-pathological entities produce thickening of the pachymeninges (Table I), hence, idiopathic hypertrophic cranial pachymeningitis (IHCPM) is a diagnosis of exclusion. A dural biopsy is essential to establish the diagnosis of hypertrophic cranial pachymeningitis and to exclude known causes. Only 33 cases of IHCPM were identified in a search of the literature till 1997.5 Goyal et al,6 reported four patients with idiopathic hypertrophic cranial pachymeningitis from India; in only two of them the diagnosis was confirmed by a meningeal biopsy. We document four more patients with biopsy proven IHCPM to elucidate the clinical, radiological and pathological features of this rare disorder. Case Report The following four patients were treated in the Neurology services of the Sree Chitra Tirunal Insitute for Medical Sciences and Technology, Trivandrum, Kerala, which is a tertiary referral center for neurological disorders, in southern part of India. Case 1 : A 52 year old women had progressive daily headache, hearing impairment, loss of smell, visual deterioration, and dysphagia for one year. She had earlier received a brief course of oral prednisolone, which resulted in a partial amelioration of her symptoms. On clinical examination, the visual acuity in the left eye was 6/12 and she could only perceive light in the right eye. Fundus showed bilateral optic atrophy. In addition, she had right III, IV, and VI, bilateral VII, and left IX, and X cranial nerve palsies. The motor and sensory system examination was normal and there were no cerebellar signs. Routine urine analysis, blood counts and serum chemistry were normal, except for an elevated erythrocyte sedimentation rate (ESR) of 55 mm/hr. The serum was negative for rheumatoid factor, antinuclear antibodies, VDRL test, hepatitis B surface antigen (HBsAg) and anti-double stranded DNA. Work up for sarcoidosis such as chest radiograph, serum calcium and angiotensin converting enzyme were negative. Bone marrow examination and ultrasound scan of the abdomen, to exclude a neoplastic and chronic inflammatory processes, were normal. Cerebrospinal fluid (CSF) analysis after lumbar puncture revealed 2 lymphocytes/mm,3 protein of 26 mg/dl and sugar of 78 mg/dl. Microbiological evaluation of the CSF for cryptococcal antigen, VDRL, and cultures for acid fast bacilli (AFB) and fungi yielded no positive results. Magnetic resonance imaging (MRI) of the brain showed thickening of the meninges which was more marked along the falx and tentorial edge. Gadolinium enhanced T1WI MRI images showed thickened uniformly enhanced meninges (Fig. 1). There were no brain parenchymal lesions. The patient underwent a left parieto-occipital craniectomy. The underlying dura was observed to be thickened, grey and grossly fibrotic. A biopsy was obtained from the right parietal dura mater. Histopathological examination of the meningeal biopsy revealed a dense fibrous tissue infiltrated with plasma cells and lymphocytes. There was no granuloma formation or vasculitis. Specific stains for fungi and acid fast bacilli were negative. She was started on oral prednisolone, 40 mg/day, which was tapered to 20 mg/day after six months. When last seen. The headache and most of her cranial nerve palsies had completely resolved by 11 months after the initiation of steroid therapy. However, the profound vision loss in the right eye remained unchanged. Case 2 : A 38 year old woman, who had hypertension and daily headache for one year, was hospitalized for a recent exacerbation of the headache. She had bifrontal, nonpulsatile, daily headache of moderate intensity, which was not associated with phonophobia and photophobia. She had no other neurological symptom. The blood pressure at admission was 130/90 mmHg. Laboratory evaluation revealed a normal blood counts, ESR and chemistry. The lumbar CSF study showed a clear fluid under normal pressure with 2 lymphocytes/mm3, protein of 96 mg/dl and sugar of 58 mg/dl. Examination of the CSF for mycobacteria, fungi and malignant cells was negative. Work up for sarcoidosis, collagen vascular diseases and systemic malignancy was negative. MRI brain showed diffuse thickening and gadolinium enhancement of dura of the falx, cranial convexity and tentorium. A meningeal biopsy taken through a left precoronal frontal burr hole showed a dense collagenous tissue with few inflammatory cells comprising lymphocytes and foci of nonspecific neovascularization. No microorganisms, granulomas or evidence of vasculitis were seen in the biopsy specimen. The patient was started on oral prednisolone 40 mg/day. Six months later, she was readmitted with recurrent episodes of left focal seizures and left hemiparesis. Her headache had completely resolved. A blood sugar of 363 mg/dl was detected. Her blood pressure was controlled on amlodipine. A repeat cranial MRI revealed multiple lacunar infarcts in the centrum semiovale and an increase in the thickening of meninges. The hyperglycemia was corrected with insulin, and treatment with diphenylhydantoin was initiated. Her left hemiparesis had partially improved by 18 months and she was still receiving a maintenance dose of 20 mg prednisolone per day. She had no further headache or seizures. Case 3 : This 30 year old man started developing recurrent episodes of neurological dysfunction at the age of 16 years, in 1985. He first presented with headache, diplopia and left sided hearing loss of three months duration. Examination confirmed left lateral rectus palsy and a left sensory-neural hearing loss. CT scan, cerebral angiogram and CSF examination were normal. Investigations for sarcoidosis, collagen vascular diseases and systemic neoplasms did not yield any positive results. He was treated with oral prednisolone for six weeks, which resulted in total resolution of his symptoms and signs. One year later he developed recurrence of headache and decrease in the vision of the right eye. These symptoms completely resolved after restarting prednisolone. Three years after the onset of symptoms, he developed recurrent right sensorimotor simple partial seizures, which responded to treatment with phenytoin. In October 1999, nearly 14 years after the onset of his remitting and relapsing neurological disorder, he had progressive bilateral vision loss and unsteadiness of gait of two weeks duration. On examination, he only had light perception in both the eyes and there was bilateral optic atrophy. The reflexes were normal, and he had gait ataxia. The only abnormal finding on routine laboratory evaluation was an elevated ESR of 50 mm/hr. The lumbar CSF study showed a protein level of 42 mg/dl with 2 lymphocytes/mm3 and normal sugar content. Examination of the CSF for AFB, fungi and malignant cells was negative. MRI brain showed thickening of dura mater of the falx and tentorium. The optic nerves were small and atrophic. The sagittal sinus was narrow secondary to dural thickening. On contrast administration, uniform enhancement of the thickened meninges and a right parietal infarct was seen. Through a right frontal craniotomy, biopsy of the anterior falx was done which showed dense fibrous tissue with inflammatory cells consisting of lymphocytes and occasional neutrophils. No granulomas or evidence of vasculitis were seen. In view of the profound and rapid deterioration of vision, he was administered a five-day course of intravenous methylprednisolone 1 gm/day, after which he was maintained on oral prednisolone 40 mg/day. At last follow-up, after three months of steroid treatment, he showed no improvement in the visual acuity, but had not developed any additional neurologic symptoms. Case 4 : A 38 year old lady was admitted with chronic headache of two years duration. Two months after the onset of headache, she was investigated elsewhere. CT scan of the head had shown tentorial enhancement and CSF examination revealed 10 lymphocytes/mm3 with normal protein and sugar values. The CSF was normal. She received antituberculous treatment for a period of nine months. Because of persistent headache, she was referred to us. Clinical examination was normal. Investigations for tuberculosis, sarcoidosis and collagen vascular diseases were negative. A CT head showed thickened enhanced tentorial meninges with dilated third and lateral ventricles with normal fourth ventricle, suggesting aqueductal obstructive hydrocephalus. The CSF study was normal. She was treated with ventriculoperitoneal shunt after which her headache subsided. It was decided to keep her under follow-up and steroids were not started. Seventeen months later, she presented with two months history of headache, dysphagia, stiffness of the lower limbs and unsteadiness of gait. On examination she had bilateral VI, IX, X and XII cranial nerve palsies, and bilateral pyramidal and cerebellar signs. The cranial MRI showed diffuse thickening and enhancement of the meninges along the floor of the anterior, middle and posterior cranial fossa and extending inferiorly to the cervical spinal canal upto the fourth cervical vertebral level (Fig. 2a and b). The spinal cord was compressed and thinned out at the second and third cervical vertebral levels. She underwent suboccipital craniectomy with a high cervical laminectomy and biopsy of the dura mater. Histopathological examination showed dense collagen tissue with collection of lymphocytes, plasma cells and few eosinophils and macrophages (Fig. 3a and b) There was no evidence of granuloma or vasculitis. She was treated with a three-day course of methyl prednisolone 1gm/day followed by maintenance of oral prednisolone 60 mg/day. During the two weeks of treatment, her dysphagia and ataxia had improved. Discussion Hypertrophic pachymeningitis is a rare disorder of diverse etiology (Table I). Charcot and Joffroy1 first described it in relation to spinal meninges. Cranial hypertrophic pachymeningitis is rarer than the spinal form. Early reports of cranial hypertrophic pachymeningitis were due to tuberculosis or syphilis.2,3 Naffziger and Stern4 described the first case of idiopathic hypertrophic cranial pachymeningitis. Since then, till 1997, 33 patients with hypertrophic cranial pachymeningitis have been documented.5 To our knowledge, four patients, two biopsy proven, reported by Goyal et al,6 appear to be the only documentation of idiopathic hypertrophic cranial pachymeningitis from India. We report four more biopsy confirmed south Indian patients, with IHCPM. Clinical features : Parney et al5 reported headache, cranial nerve palsy and ataxia in 88%, 62% and 32% of the cases, respectively. Headache may be the only symptom for many years in a small percentage of patients.7 Cranial nerve palsies are due to compression of the exit zone of nerve roots by the hypertrophic basal pachymeningitis.8 The eighth cranial nerve is most frequently involved, followed in equal frequency by the optic, ocular and lower cranial nerves.5,9 Papilledema was noted among 9 of 33 patients (26%).5 Ischemia caused by tight, thickened and adherent pachymeninges of the posterior fossa is presumed to be responsible for the cerebellar dysfunction.9 There are isolated reports of dural venous sinus occlusion,6,10,11 internal carotid artery occlusion,12 intracranial hemorrhage26 and obstructive hydrocephalus13,14 among patients with IHCPM. Age ranges from 20 to 78 years (mean 51 years).5 Three of our four patients (mean age 40 years, range 30 to 52 years) were females. All our patients presented with chronic headache. While three of them had multiple cranial nerve palsies, gait disturbance related to cervical myelopathy dominated in patient no. 4. Two of our patients (patients 1 and 3) had profound irreversible vision loss. Symptomatic cervical hypertrophic pachymeningitis as observed in patient no. 4, and cerebral parenchymal infarcts and focal seizures as noted in patients no. 2 and 3 in association with idiopathic hypertrophic cranial pachymeningitis is seldom documented in the literature.9,13 Diagnosis : A variety of infectious, inflammatory, infiltrative, neoplastic and other disease processes can cause diffuse thickening of the dura mater (Table I). Varied clinical presentations of these disorders and the difficulty in establishing the etiology can lead to diagnostic dilemmas. IHCPM is a diagnosis of exclusion. An elevated ESR was present in 24 out of 27 patients with hypertrophic cranial pachymeningitis. 5 Three of our patients had an ESR of over 50 mm/1h. The CSF protein may be elevated in twothird, lymphocytic pleocytosis may occur in onefourth, and the CSF may be normal in one-fourth of patients with hypertrophic cranial pachymeningitis.5 The CSF was normal in our patients, except for an elevated protein in patient no. 2. IHCPM is being increasingly recognized with the advent of CT and MRI. MRI is superior to CT in the diagnosis of hypertrophic cranial pachymeningitis. Unenhanced CT shows thickened hyperdense dura, typically involving the tentorium, falx, and prepontine region, which enhances markedly after contrast administration. On MRI, the thickened dura mater appears isointense or hypointense on T1WI and hypointense on T2WI often associated with a hyperintense edge.15 Thickening is better appreciated in the coronal and sagittal images. Contrast administration reveals uniform enhancement of the thickened meninges. The low MRI signal represents dense fibrosis, and enhancement suggests inflammation. Rarely, nodular pseudotumoral thickening of dura mater, mimicking multiple meningioma may occur.16 A meningeal biopsy is usually required to establish the diagnosis of IHCPM and to exclude other causes of hypertrophic pachymeningitis. Pathological findings consist of thick fibrous dura often associated with chronic inflammatory cell infiltrate consisting of lymphocytes and plasma cells. Granulomatous findings have been noted in about 10% of the reported cases of hypertrophic cranial pachymeningitis.9,17,18 Biopsy from an accessible site with CT or MRI documented enhancing and thickened dura mater is more likely to yield a positive etiological diagnosis.19 The varied meningeal biopsy sites in our patients are due to our practice of selecting the optimal site of biopsy based on neuroimaging findings. In developing countries, a majority of patients presenting with clinical and investigative features suggestive of a chronic meningitic process will appropriately receive a trial of antituberculous chemotherapy before alternate diagnostic possibilities are considered, as in our patient no. 4. Tuberculous meningitis is a diagnosis very difficult to exclude. Parney et al5 described a patient with hypertrophic pachymeningitis, who responded to antituberculous therapy in whom exhaustive investigations, including a meningeal biopsy, failed to establish an underlying cause. Polymerase chain reaction (PCR) of CSF may be helpful in such cases. We recently reported a case in whom a diagnosis of en-plaque meningeal tuberculoma based on MRI findings was confirmed by a Mycobacterium tuberculosis complex-specific PCR assay of the CSF.20 Treatment : The optimal treatment of IHCPM is unknown. Spontaneous resolution of both clinical symptoms and signs, and dural thickening has been reported.21 A variety of therapeutic approaches have been tried alone or in combination. Corticosteroid therapy is often effective in ameliorating the symptoms and signs, and in arresting the progress of the disease.8,13,22,23 In some patients, serial imaging studies have shown a reduction in the thickness and degree of enhancement of the meninges.9 Rarely patients have worsened while on steroid therapy.6 In those who do not respond to steroids or those who develop steroid dependence with attended side effects, cyclophosphamide and azathioprine have been tried with benefit.9,13,16 Surgical excision is an option for patients with mass effect due to thickening of skull base dura, unresponsive to steroid therapy.9 Symptomatic hydrocephalus requires ventriculoperitoneal shunting as in our patient no. 4. Decompression of the optic nerve has resulted in dramatic improvement in a patient with rapidly deteriorating vision.24 Natural history : The natural course of idiopathic hypertrophic cranial pachymeningitis is poorly understood. Spontaneous resolution, response to steroids, steroid dependency, and remitting and relapsing course have been documented.9,13,21 Among the 33 patients with IHCPM gathered from the literature by Parney et al,5 26% experienced full remission without steroid dependence, 15% experienced steroid dependent partial or complete remission, 15% experienced progressive course inspite of steroid therapy, and 32% died regardless of treatment. In retrospect, our initial decision to withhold steroid therapy for patient no. 4, when her headache ameliorated after ventriculo-peritoneal shunt, was inappropriate. Within the next two years, she developed neurologic deterioration due to progression of the pachymeningitic process. Early institution and long-term maintenance of steroid therapy may prevent the neurologic sequelae associated with idiopathic hypertrophic cranial pachymeningitis. Etiopathogenesis : Idiopathic hypertrophic cranial pachymeningitis is histologically similar to its spinal counterpart and they are presumed to be different presentations of a single disease. As noted in patient no. 4, these two entities may overlap. There are clinical and pathological similarities between IHCPM, Tolosa-Hunt syndrome, polyneuritis cranialis, orbital pseudotumor and multifocal fibrosclerosis.9,25 Clinically, all these disorders can result in pachymeningitis with associated overlap in the symptoms and signs. All have a tendency to respond to corticosteroids. Chronic nonspecific noncaseous inflammatory processes characterizes all. While Tolosa-Hunt syndrome may be a focal manifestation of pachymeningitic process involving the walls of the cavernous sinuses, IHCPM may be a localized manifestation of multifocal fibrosclerosis. Although an immunological basis is suspected, the etiopathogenesis of this group of chronic fibrosing inflammatory disorders at present unknown. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02013f3.jpg] [ni02013f2.jpg] [ni02013t1.jpg] [ni02013f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}