 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
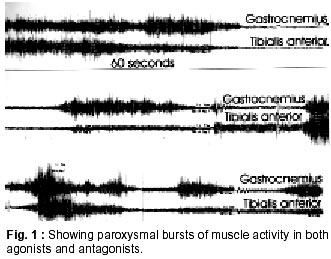
Neurology India, Vol. 50, No. 1, March, 2002, pp. 98-99 SHORT REPORT Stiff Person Syndrome and Myasthenia Gravis P.K. Saravanan, J. Paul, Zaheer Ahmed Sayeed Correspondence to : Zaheer Ahmed Sayeed, GI, Palmwoods, 9 Seshadri Road, Chennai - 600 018, India. Code Number: ni02026 Summary Association of stiff person syndrome, an immune related disorder of anterior horn cells and myasthenia gravis an endplate disorder with similar pathogenesis, is rare. This communication documents this association in the Indian literature for the first time. Key words : Stiff person syndrome, Myasthenia gravis. Introduction Stiff person syndrome (SPS) is a rare disorder characterized by muscle rigidity, spasms and continuous motor unit activity. Initially described by Moersch and Woltman in 1956,1 SPS has emerged to be a treatable dysfunction of the anterior horn cells and should be a consideration in patients with unexplained stiffness and spasms. SPS begins insidiously, initially with involvement of axial musculature, and spreads to the limb muscles. Occasionally any one of the limb muscles may be involved initially. The pathogenesis of SPS has been better defined. These include demonstration of i) antibodies against glutamic acid decarboxylase (GAD), a rate limiting enzyme for the synthesis of inhibitory neurotransmitter d -amino butyric acid in the presence of various other autoantibodies and ii) the association with other disimmune pathological processes e.g. diabetes mellitus, thyrotoxicosis, pernicious anemia, and vitiligo. This communication documents the association of SPS with myasthenia gravis (MG), a disorder consequent to abnormal immune response directed to b subunit of the Ach receptors, an infrequent combination! Case Report A 43 year old lady presented with chronic pain and stiffness in the low back and both lower limbs. These were initially noticed following cesarean section 15 years ago. For several years since 1984, she had been experiencing intermittent painful spasms involving the lower extremities which were treated with analgesics, without significant relief. Except for involvement of paraspinal muscles of the back and lower thoracic region, the upper limbs remained uninvolved. Fifteen years earlier, she underwent surgical correction of squint. The details of the surgery on the right eye were not available for evaluation. Since then, ptosis involving both eyes independently and intermittently at different occasions, characterized the clinical course of her complaints. Clinical examination showed normal vital signs, with a BP of 130/80 mmHg. No significant skin abnormalities were obvious. Neurological examination revealed mild bilateral ptosis exacerbated by persistent upward gaze, restriction of upward conjugate gaze with worsening on continued looking up, restriction of lateral conjugate gaze on looking persistently to either direction, reduction in power of both grips on quick alternate gripping and relaxing over a period of five minutes. There was no difficulty with deglutition, or neck and proximal muscle weakness. She was able to sit and get up from ground without help. During the neurological interview and examination, several episodes of painful contraction of gluteal, limb and back muscles were noted, which involved both agonist and antagonist groups of muscles, in the lower extremities with similar involvement of paraspinal and abdominal musculature. The sensory and cerebellar systems showed no abnormalities. The deep tendon reflexes were normal. Laboratory investigations showed normal hemogram, biochemical parameters and distribution pattern of serum proteins. Imaging of the lumbosacral spine and chest showed no significant abnormalities. Electro-physiology documented paroxysmal bursts of muscle fibre activity of high amplitude in the agonist and antagonist group of muscles simultaneously during the period of muscle spasms flowing over periods of relaxations as well (Fig. 1). Intravenous prostigmine (1 mg) reversed the decremental response of the ocular, facial and skeletal muscle. Values of anti-AchR antibodies and anti-GAD antibodies could not be documented. Painful muscle spasms abated with intravenous diazepam. Prostigmine remedied the post synaptic end plate defect. The patient refused immunosuppressive therapy. Discussion Immunogenesis of SPS was identified in 1988 with the report of autoantibodies to GAD-65. Antibodies against the two isoforms of GAD-65 and GAD-67 are present in about 68% of patients with SPS.2 GAD is also present in beta cells of pancreas and 30% of patients with SPS show a positive association with diabetes mellitus. Other autoantibodies found in association with SPS include antithyroid antibodies, anti parietal cell antibodies, antinuclear antibodies, antibodies against JO-I antigen and ribonucleoprotein. SPS manifesting as remote effect of neoplasia exhibits antibodies against amphysin, and neuronal proteins localized in neurons and synaptic vesicles. Such antibodies are also seen in patients with myasthenia gravis without manifest neoplasia. Immunogenetic studies in SPS show a high frequency of association with HLA-DR or DQ haplotypes particularly with alleles in the DR beta I and DQ beta 1 locus.3 The association of SPS and MG has been reported previously. MG has preceded SPS,4 but is a rare association, not serendipitous, with diabetes mellitus and inflammatory thyroid disease. Ocular MG and symptoms of SPS were relieved following thymectomy for histologically proven thymoma.5 Generalized MG developed six years following spontaneous remission of SPS, again in association with a thymoma.6 Besides antibodies to acetylcholine receptor, the anti-nuclear, anti-DNA, antimitochondrial and anti skeletal muscle antibodies have also been demonstrated in patients. The diagnosis of neuromuscular hyperactivity syndrome underwrites a vigorous search directed towards an exclusion of a thymic enlargement secondarily to a thymoma. Thymic enlargement in the patient under review was absent. Chimeric monoclonal antibody has been known to provide relief from symptoms of MG.7 MG and SPS represent a dysfunction of the two ends of the lower motor neuron, albeit with different chemical immunogenic antigens. Use of Chimeric monoclonal CD4 antibody in SPS has not been documented in literature, as in other altered immune states. The combination of MG and SPS occurring in the same individual deserves such a trial and, if successful, could suggest the unique commonality of the gene locus in these diverse disorders. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02026f1.jpg] |
| |||||||||

{kind=link}