 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
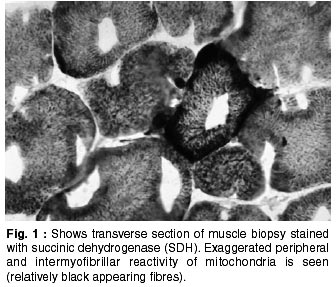
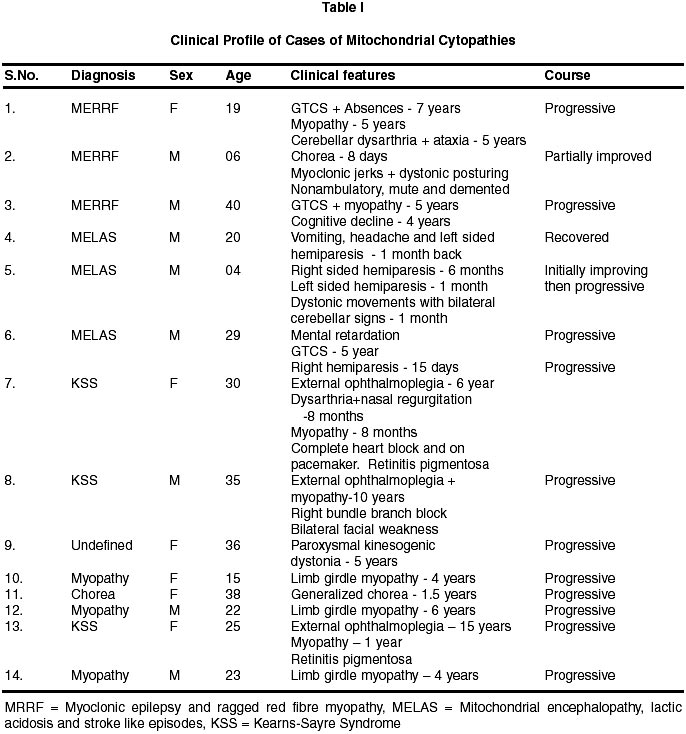
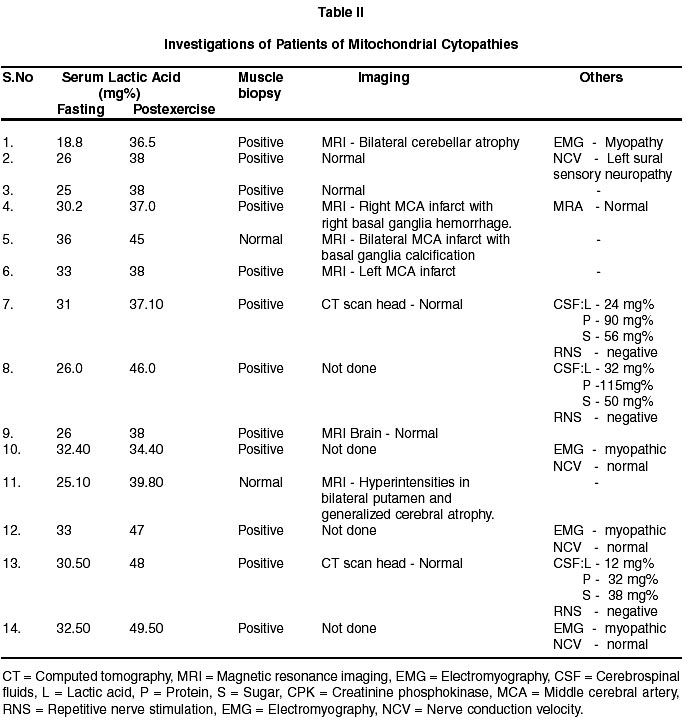
Neurology India, Vol. 50, No. 2, June, 2002, pp. 162-167 Neurological Mitochondrial Cytopathies M.M. Mehndiratta, P. Agarwal, M. Tatke,* M. Krishnamurthy Departments of Neurology and Pathology*,
G.B. Pant Hospital,
New Delhi-110002, India. Accepted for publication : 9th July, 2001 Code Number: ni02046 Summary The mitochondrial cytopathies are genetically and phenotypically heterogeneous group of disorders caused by structural and functional abnormalities in mitochondria. To the best of our knowledge, there are very few studies published from India till date. Selected and confirmed fourteen cases of neurological mitochondrial cytopathies with different clinical syndromes admitted between 1997 and 2000 are being reported. There were 8 male and 6 female patients. The mean age was 24.42±11.18 years (range 4-40 years). Twelve patients could be categorized into well-defined syndromes, while two belonged to undefined group. In the defined syndrome categories, three patients had MELAS (mitochondrial encephalopathy, lactic acidosis and stroke like episodes), three had MERRF (myoclonic epilepsy and ragged red fibre myopathy), three cases had KSS (Kearns-Sayre Syndrome) and three were diagnosed to be suffering from mitochondrial myopathy. In the uncategorized group, one case presented with paroxysmal kinesogenic dystonia and the other manifested with generalized chorea alone. Serum lactic acid level was significantly increased in all the patients (fasting 28.96±4.59 mg%, post exercise 41.02±4.93 mg%). Muscle biopsy was done in all cases. Succinic dehydrogenase staining of muscle tissue showed subsarcolemmal accumulation of mitochondria in 12 cases. Mitochondrial DNA study could be performed in one case only and it did not reveal any mutation at nucleotides 3243 and 8344. MRI brain showed multiple infarcts in MELAS, hyperintensities in putaminal areas in chorea and bilateral cerebellar atrophy in MERRF. Key words : Mitochondrial cytopathies, MELAS, MERRF, KSS, Mitochondrial myopathy. Introduction The mitochondrial cytopathies are genetically and phenotypically heterogeneous group of disorders caused by structural and functional abnormalities in mitochondria leading to involvement of nervous system (mitochondrial encephalomyopathies) and other organ systems (mitochondrial cytopathies).1,2 It can present at any time from infancy to late adulthood.3 Abnormalities of the electron transport and oxidative phosphorylation (oxphos) system are probably the most common causes of mitochondrial cytopathies.3 Over the past decade, a large body of evidence has accumulated implicating defects of human mitochondrial DNA in the pathogenesis of a group of disorders known collectively as the mitochondrial encephalomyopathies.4 Neurological manifestations of mitochondrial cytopathies encompass a diverse group of clinical manifestations including certain defined clinical syndromes and multisystem manifestations.5 There are very few studies of neurological mitochondrial cytopathies from India.6-8 Fourteen cases of neurological mitochondrial cytopathies presenting with different types of clinical features are being reported. Material and Methods Fourteen cases were diagnosed to be suffering from neurological mitochondrial cytopathies between 1997 and 2000 in the department of Neurology, G.B. Pant Hospital, New Delhi. All the cases were investigated thoroughly to rule out other etiologies with relevant investigations. Antistreptolysin O (ASO) titre was less than 200 IU and CRP was negative in cases of chorea. Patients presenting with childhood and young stroke were investigated to rule out common causes of young stroke. There was no evidence of cardiac source of embolization and antiphospholipid antibody was negative in cases of MELAS presenting as young stroke. Serum lactic acid (fasting and post exercise) was estimated in all the cases through enzymatic determination, by colorimetric method, using kit of Randox Laboratories Ltd. U.K. The final diagnosis for labelling the cases of neurological mitochondrial cytopathies was based on diagnostic criteria described by Walker et al in 1996.5 The muscle biopsy was taken from quadriceps in all cases, under proper aseptic technique. All biopsy specimens were stained with hematoxylin and eosin (H&E), succinic dehydrogenase (SDH), and Gomori-trichrome (GMT) to evaluate the architecture of muscle, fibre types and mitochondrial abnormalities. Increased intramyofibrillar peripheral enzyme reactivity due to overproduction of abnormal mitochondria was seen as purplish structure at the periphery of the myofibrils on SDH stain (Fig. 1) and ragged red fibres on GMT stain. These mitochondrial abnormalities are the hallmark of mitochondrial cytopathy.9,10 Electron microscopic examination of muscle biopsy was also done in six cases to find the evidence of proliferation of abnormal mitochondria. The genetic analysis could only be done in one patient of MERRF. All patients were treated with high doses of vitamins (thiamin, riboflavin, vitamin C, K) antioxidants (vitamin E) and other supportive measures. CoQ10 was prescribed to only one patient because of non-availability of this product in the Indian market. Results In the present study, there were 8 male and 6 female patients. The mean age of presentation was 24.42±11.18 years (range 4-40 years). Myoclonic epilepsy with ragged red fibre myopathy (MERRF), mitochondrial encephalopathy, lactic acidosis and stroke like episodes (MELAS), Kearns-Sayre syndrome (KSS) and mitochondrial myopathy were diagnosed in 3 cases each. One case each had paroxysmal kinesogenic dystonia and generalized chorea (Table I). All the cases were investigated to rule out other etiologies according to their clinical presentations and to confirm the diagnosis of mitochondrial cytopathy. Hemogram, ESR, urine analysis, renal function tests, liver function tests, and X-rays chest were within normal limits in all. Different investigations were done in each patient depending on the mode of presentation, to rule out other possibilities. Repetitive nerve stimulation (RNS) was negative in patients of KSS. Electromyography (EMG) was suggestive of myopathy in patients of mitochondrial myopathy. The serum lactic acid was significantly increased (fasting 28.96±4.59 and post exercise 40.87±5.04 mg%) in all cases as compared to the controls (4.5-20 mg%). SDH staining of muscle biopsy showed subsarcolemmal accumulation of abnormal mitochondria in 12 cases and 3-40% red ragged fibres on GMT staining in ten cases, while muscle biopsy was normal in the remaining two cases (Table II). The electron microscopic examination showed evidence of abnormal mitochondria (inclusions, giant mitochondria) or proliferation of mitochondria, normal in structure and size, in six cases which further aided the diagnosis. All the cases had negative family history, except in one case of paroxysmal kinesogenic dystonia in which the patient's daughter had mental retardation. MRI brain showed multiple infarcts in patients of MELAS, hyper intensities in putaminal areas in the patient presenting solely with chorea and bilateral cerebellar atrophy in MERRF. Mitochondrial DNA study, performed by PCR endonuclease digestion, did not reveal any mutation at nucleotides 3243 and 8344 in a patient of MERRF. Discussion The term mitochondrial encephalomyopathy was first used by Shapira in 1977 to describe cases with complex multisystem disease with structurally and/or functionally abnormal mitochondria in brain or muscles.11 Initially mitochondrial cytopathies were regarded as rare clinical entities, but in the last few years due to heightened awareness and research, mitochondrial abnormalities have been attributed in many neurological disorders. Neurological manifestations of mitochondrial cytopathies comprise of multineuraxial involvement with raised serum lactic acid and positive muscle biopsy for mitochondrial abnormalities confirmed by genetic analysis for mitochondrial DNA/nuclear DNA mutation. Occasional ragged red fibres may be seen in other muscle disoders like muscular dystrophy and polymyositis as well. Clinical correlation with investigations need to be done, to give a label of mitochondrial disorders. MERRF is characterized by mitochondrial myopathy with various seizures types (myoclonic epilepsy, generalized tonic-clonic seizure, focal seizures) and variable central nervous system (CNS) dysfunctions ranging from severe CNS dysfunction (deafness, ataxia, spasticity, myoclonus, dementia, pigmentary retinopathy), cardiomyopathy, renal tubular dysfunction and peripheral neuropathy to asymptomatic myopathy with red ragged fibres.7,12,13 Our patients had most of the classical features of MERRF in the form of seizures (myoclonic seizure in 2, generalised seizures in 2 and absence seizure in 1 case, myopathy in all the three, cerebellar ataxia in one, dementia in two, raised serum lactic acid in all the three and red ragged fibres in muscle biopsy in all the three cases (Tables I, II). Mitochondrial DNA study performed by PCR endonuclease digestion did not reveal any mutation at nucleotides 3243 and 8344, while mutation at nucleotide 8356 was not tested. The lack of mutation on testing is due to genetic heterogeneity present in patients of MERRF. The point mutation has been found at multiple sites e.g. nucleotides 3243 or 8344 or 835614 in transfer RNA or mitochondrial DNA gene. All the three patients of MERRF fall in the definitive group of mitochondrial cytopathies according by Walker et al.5 One patient showed gradual improvement on high doses of vitamins (thiamin, riboflavin, vitamin C and K) and antioxidants (vitamin E) with avoidance of phenytoin and barbiturates. He started walking and vocalizing. His generalised tonic clonic seizures were controlled, although brief myoclonic jerks were persisting. MELAS was first described by Pavlakis et al15 in 1984. It is characterized by normal early development, followed by onset of exercise intolerance, stroke like episodes, seizures and dementia. In a large series reviewed by Hirano et al,16 all patients became symptomatic before the age of 40 years and had evidence of lactic acidosis. The skeletal muscle biopsy revealed RRF. Three cases of MELAS reported here had onset of symptoms at young age (4-29 years) with similar symptoms (Table I). They had stroke like episodes in middle cerebral artery territory. One patient had GTCS and one had headache and vomiting. One third of the patients can have basal ganglia calcification. This manifestation was observed in one of our patients. Muscle biopsy was positive for mitochondrial cytopathy (SDH and GMT staining) in two patients (Table II). Muscle biopsy can be negative for mitochondrial cytopathy in patients of MELAS,17 as it was in two of our cases. However, lactic acidosis was present in all patients. The family history was not contributory, however, genetic analysis was not done because patients could not afford the same. Two patients fulfilled two major criteria and hence fell in the definitive group. One patient fulfilled one major and one minor criterion, hence, was included in the probable group of mitochondrial cytopathy on the basis of Walker's criteria.5 One patient showed complete recovery and the other showed partial recovery with high doses of vitamins (thiamin, riboflavin, C, K) and antioxidants, on follow up. KSS is characterized by progressive external opthalmoplegia, cardiac conduction block, pigmentary retinal degeneration, dementia, CSF protein > 100 mg/dl and variable number of red ragged fibres on muscle biopsy. Other features include deafness, ataxia, episodic coma and endocrinal abnormalities.18 All the three patients of KSS had progressive external opthalmoplegia. Two had pigmentary retinal degeneration and cardiac conduction defect, and one had myopathy. One patient had bulbar symptoms in the form of dysarthria and nasal regurgitation while the other had bilateral facial weakness, which are infrequent findings in these patients. Serum lactic acid was significantly raised and muscle biopsy showed red ragged fibres in all the three patients. CSF lactate was increased in all three and protein was increased (90 and 115 mg%) in two patients. All the three patients fulfilled two major criteria, therefore fell in the definitive group of mitochondrial cytopathy. The patients of mitochondrial myopathy were young (age 15-23 years). They had gradually progressive muscle weakness of four to six years duration without any other system involvement. The metabolic parameters (serum calcium, sugar) and hormone levels (thyroid, parathyroid) were normal. EMG showed myopathic pattern and NCV was normal in all three patients. The serum lactic acid was significantly increased and muscle biopsy (SDH and GMT staining) was positive for mitochondrial cytopathy in all the patients. These patients had one major (positive muscle biopsy) and one minor criteria (raised serum lactate) for diagnosis and therefore were included in probable group of mitochondrial myopathy. Movement disorders have been described due to mitochondrial abnormalities.19,20 Dystonia has been the most frequent movement disorder20 besides myoclonus, chorea, athetosis, and tremors. Mitochondrial abnormalities have been found even in Huntington's disease and Parkinson's disease.19 In the present series, two young (36 and 38 years) patients had slowly progressive paroxysmal kinesogenic dystonia and generalized chorea respectively. MRI brain was normal in one while hyper-intensities in bilateral putamen and generalized cerebral atrophy were present in the other. The serum lactic acid was significantly increased in both. Muscle biopsy was positive for mitochondrial cytopathy in one patient. These patients were regarded as probable mitochondrial cytopathy, which can be confirmed, by biochemical or polarographic assessment of respiratory chain complexes or genetic analysis. We still don't have the facility for analysis of respiratory chain analysis. Treatment options for patients of mitochondrial cytopathy are limited. Dietary manipulations including high carbohydrate diet have been recommended to compensate for impaired gluconeogenesis and to decrease lipolysis. The patients should avoid extremes of temperature and drugs like phenytoin and barbiturates.7 Anecdotal success has been reported with a number of vitamins and co-factors (thiamin, riboflavin, vitamin C and K, carnitine) and glucocorticoids.21,22 Coenzyme Q10 has been found to be consistently beneficial in some studies,23,24 and has been marketed in India since January 2001. The prognosis is variable, as the disease may have fluctuating course. Mortality is usually due to cardiac conduction defects. In conclusion, the patients of mitochondrial cytopathy described here were of young age and had multisystem involvement with significantly increased serum lactic acid. SDH staining was positive in muscle biopsy in most of the cases (85.71%). Therefore, patients with multi-neuroaxial involvement should also be investigated on the lines of mitochondrial cytopathies, if the symptoms complex cannot be explained by defined known disorders. Though serum lactic acid is not considered to be a specific test, however, it can provide a lead to the diagnosis, specifically when significantly raised. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02046t2.jpg] [ni02046t1.jpg] [ni02046f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}