 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
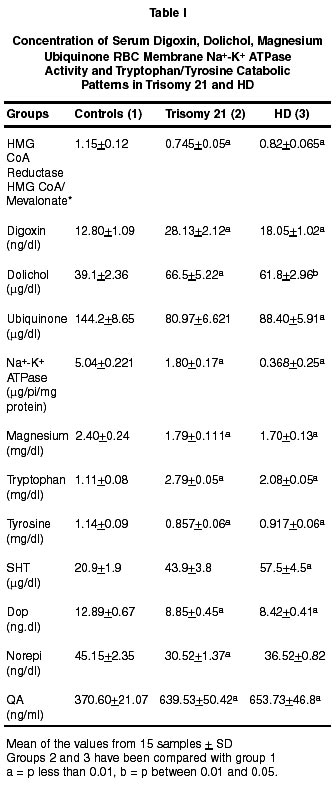
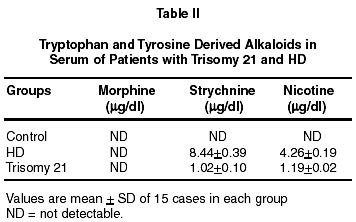
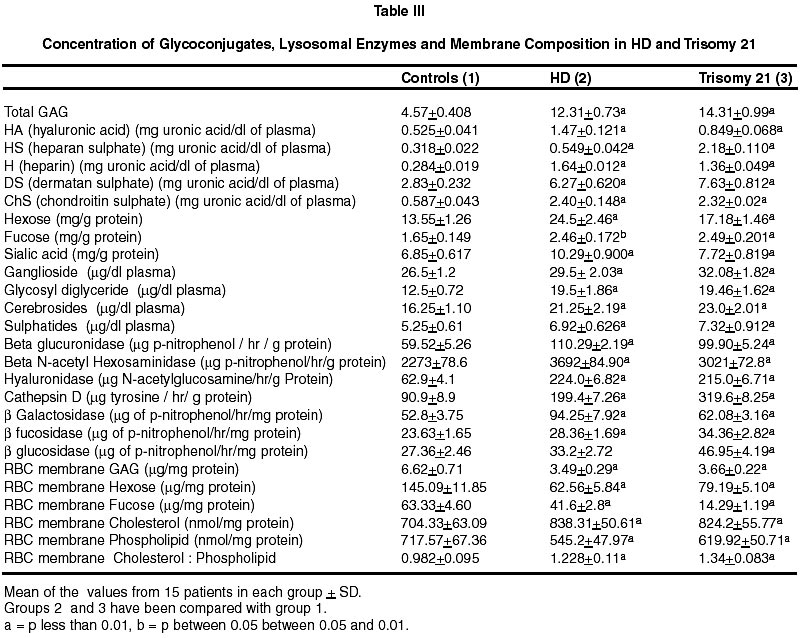
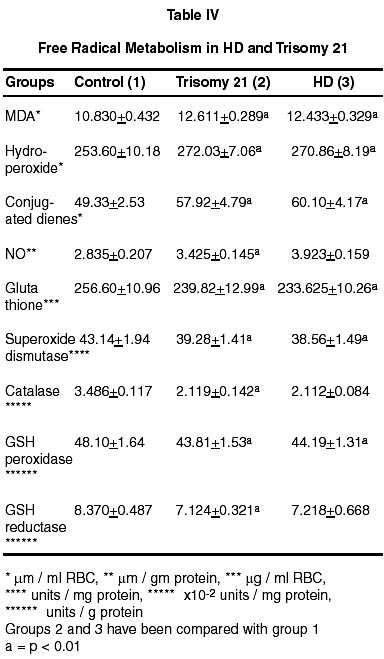
Neurology India, Vol. 50, No. 2, June, 2002, pp. 174-180 Endogenous Sodium-Potassium ATPase Inhibition Related Biochemical Cascade in Trisomy 21 and Huntington's Disease : Neural Regulation of Genomic Function A. Ravi Kumar, P.A. Kurup* Department of Neurology,
Medical College Hospital,
and
Department of Biochemistry*,
University of Kerala,
Trivandrum, Kerala, India. Accepted for publication : 6th June, 2001 Code Number: ni02048 Summary The isoprenoid pathway related cascade was assessed in trisomy 21 and Huntington's disease. Membrane Na+-K+ ATPase activity, serum magnesium and ubiquinone were decreased while HMG CoA reductase activity, serum digoxin and dolichol levels were increased in both the disorders. There were increased levels of tryptophan catabolites (nicotine, strychnine, quinolinic acid and serotonin) and decreased levels of tyrosine catabolites (dopamine, noradrenaline and morphine) in both trisomy 21 and Huntington's disease. There was an increase in dolichol levels, carbohydrate residues of glycoproteins, glycolipids, total/individual GAG fractions and lysosomal enzymes in both disorders. Reduced levels of ubiquinone, reduced glutathione and free radical scavenging enzymes as well as increased lipid peroxidation products and nitric oxide were noticed in both the disorders. The role of hypothalamic digoxin and a disordered isoprenoid pathway in the pathogenesis of trisomy 21 and Huntington's disease is discussed. Key words : Digoxin, Dolichol, Membrane Na+-K+ ATPase, Trinucleotide repeats, Chromosomal non-dysjunction, Magnesium. Introduction The pathology of the brain in trisomy 21 is akin to that in Alzheimer's disease with amyloid plaques described in both.1 Defects in the isoprenoid pathway have been described in neuronal degeneration and Alzheimer's disease.2 It has been noticed that there is an increased incidence of Alzheimer's disease in relatives of patients with trisomy 21.1 The isoprenoid pathway produces four important metabolites important in cellular function - digoxin (endogenous inhibitor of membrane Na+-K+ ATPase produced by hypothalamus), ubiquinone (a component of the mitochondrial electron transport chain), dolichol (important in N-glycosylation of proteins) and cholesterol (which is a component of cellular membrane).3,4 Alteration in ubiquinone, dolichol and cholesterol has been described in degenerative disorders like Alzheimer's disease.2 There is no data on the changes in the isoprenoid pathway in trisomy 21 and Huntington's disease (HD). Digoxin has been reported to regulate the transport of neutral aminoacids and modulate their catabolism.5 There are increased levels of tryptophan and tryptophan catabolite, quinolinic acid in Huntington's disease (HD).6 Quinolinic acid can function as a NMDA agonist contributing to NMDA excitotoxicity and neuronal degeneration. Digoxin, by altering intracellular calcium/magnesium ratios and alteration in ubiquinone levels, can affect mitochondrial function. Mitochondrial dysfunction has been described in Huntington's disease.7 A decrease in complex 1 activity of platelets has been reported in HD. The results of the study on the isoprenoid pathway related biochemical cascade in the two neurogenetic syndromes are described in this paper. Material and Methods 15 cases each of trisomy 21 and Huntington's disease (HD) attending the metabolic and genetics clinic of Medical College Hospital, Trivandrum were chosen for the study. Each patient had an age and sex matched control. All patients and controls were non-smokers (passive or active). All blood samples were collected from the patients before starting treatment. Activity of HMG CoA reductase of the serum was determined using the method of Rao and Ramakrishnan by determining the ratio of HMG CoA to mevalonate.8 For the determination of the RBC Na+-K+ ATPase activity of the erythrocyte membrane, the procedure described by Wallach and Kamat was used.9 Digoxin in the serum was determined using the procedure described by Arun et al.10 For estimation of ubiquinone and dolichol in the serum, the procedure described by Palmer et al was used.11 Magnesium in the serum was estimated by atomic absorption spectrophotometry.12 Tryptophan was estimated by the method of David and William and tyrosine by the method of Wong et al.13,14 Serotonin was estimated by the method of Curzon and Green and catecholamines by the method of Well-Malherbe.15,16 Quinolinic acid content of serum was estimated by HPLC (C18 column micro BondapakTM 4.6x150 nm), solvent system 0.01 M acetate buffer (pH 3.0) and methanol (6:4), flow rate 1.0 ml/minute and detection UV 250 nm. Morphine, strychnine and nicotine were estimated by the method described by Arun et al.17 Details of the procedures used for the estimation of total and individual GAG, carbohydrate components of glycoproteins, activity of GAG degrading enzymes and activity of glycohydrolases have been described before.18 Serum glycolipids were estimated as described in Methods in Enzymology.19 Cholesterol was estimated by using commercial kits supplied by Sigma Chemicals, USA. SOD was assayed by the method of Nishikimi et al as modified by Kakkar et al.20 Catalase activity was estimated by the method of Maehly and Chance, glutathione peroxidase by the method of Paglia and Valentine as modified by Lawrence and Burk and glutathione reductase by the method of Horn and Burns.21-23 MDA (malondialdehyde) was estimated by the method of Will and conjugated dienes and hydroperoxides by the procedure of Brien.24,25 Reduced glutathione was estimated by the method of Beutler et al.26 Nitric oxide was estimated in the plasma by the method of Gabor and Allon.27 Statistical analysis was done by student's 't' test. Results (i) The activity of serum HMG CoA reductase and the concentration of serum digoxin and dolichol were increased in HD and trisomy 21. The concentration of serum ubiquinone and magnesium, and the activity of erythrocyte membrane Na+-K+ ATPase were decreased (Table I). (ii) The concentrations of serum tryptophan, quinolinic acid and serotonin were increased in these patients while that of tyrosine, dopamine and noradrenaline were decreased (Table II). (iii) Nicotine and strychnine were detected in the serum of patients with trisomy 21 and HD but were not detectable in control serum. Morphine was not detected in the serum of these patients (Table III). (iv) The concentration of total glycosaminoglycans (GAG) was increased in the serum of HD and trisomy 21 patients. The concentration of heparan sulphate (HS) heparin (H), dermatan sulphate (DS), chondroitin sulphates (ChS) and hyaluronic acid (HA) was increased. The concentration of total hexose, fucose and sialic acid was increased in the glycoproteins of the serum in these patients. The concentration of gangliosides, glycosyl-diglycerides, cerebrosides and sulphatides showed significant increase in the serum in these patients (Table IV). (v) The activity of glycosaminoglycan (GAG) degrading enzymes-beta-glucuronidase, beta-N-acetyl hexosaminidase, hyaluronidase and cathepsin-D was increased in HD and trisomy 21 when compared to the controls. The activity of beta-galactosidase, betafucosidase and beta-glucosidase was increased in HD and trisomy 21. (vi) The concentration of total GAG and hexose and fucose residues of glycoproteins in the RBC membrane was decreased significantly in HD and trisomy 21. The concentration of RBC membrane cholesterol increased while that of phospholipid decreased. The ratio of RBC membrane cholesterol : phospholipids increased in HD and trisomy 21. (vii) The activity of superoxide dismutase (SOD), catalase, glutathione reductase and glutathione peroxidase in the erythrocytes decreased significantly in HD and trisomy 21. The concentration of malondialdehyde (MDA), hydroperoxides, conjugated dienes and nitric oxide (NO) increased significantly. The concentration of reduced glutathione decreased in HD and trisomy 21. Discussion The increase in the activity of HMG CoA reductase in trisomy 21 and HD suggests an upregulation of the isoprenoid pathway and increased digoxin synthesis. In this connection, incorporation of 14C-acetate into digoxin in rat brain has been shown by us indicating that acetyl CoA is the precursor for digoxin biosynthesis in mammals also.28 The increase in endogenous digoxin, a potent inhibitor of membrane Na+-K+ ATPase, can decrease this enzyme activity in trisomy 21 and HD. The inhibition of Na+-K+ ATPase by digoxin is known to cause an increase in intracellular calcium and reduction in intracellular magnesium stores.29 Serum magnesium was assessed in trisomy 21 and HD and was found to be reduced. DNA polymerase requires magnesium for its function. The 3' exonuclease activity of DNA polymerases 1 and III is the device for proofreading the newly made DNA strands and for correcting errors made by the polymerase activity.30 The proof reading function of the DNA polymerase is very efficient and contributes a factor of at least 104 in guaranteeing the fidelity of replication. Membrane Na+-K+ ATPase inhibition can produce intracellular magnesium depletion leading on to a defect in the proofreading function of DNA polymerase during DNA replication.30 This may possibly lead on to the genesis of trinucleotide repeats described in HD.31 Intracellular magnesium depletion can also produce defective phosphorylation of MAP (microtubule associated proteins). This results in defective microtubule related spindle fibre function and chromosomal non-dysfunction, probably contributing to Trisomy 21. The same reason holds good for the broken appearance and fragile sites of the chromosome in fragile X syndrome.31 Thus the genetic defect described in these two syndromes may partly be contributed by hypothalamic digoxin induced membrane Na+-K+ ATPase inhibition. Digoxin can possibly regulate the function of heatshock protein which functions as a molecular chaperone involved in protein folding and maturation.32 The heat-shock protein has an ATP/ADP switch domain that regulates HSP conformation. HSP is dysfunctional in the presence magnesium deficiency. Normally cellular mutations are masked by HSP90, one of the heat shock proteins. But when HSP90 is out of commission, it can no longer stabilise mutant proteins and keep them working properly. Instead the mutations are unmasked and revealed. Thus the brain can regulate genomic function by hypothalamic digoxin acting on the neuronal or cell membrane. Digoxin can regulate neutral amino acid transport. Two of the neutral aminoacids tryptophan, a precursor for strychnine and nicotine and tyrosine, a precursor for morphine are important.17,33 There is an increase in tryptophan and its depolarising catabolites (serotonin, nicotine and quinolinic acid) and a reduction in tyrosine and its hyperpolarising catabolites (dopamine, norepinephrine and morphine) in trisomy 21 and HD.34 This could be due to the fact that digoxin can preferentially promote tryptophan transport over tyrosine. Membrane Na+-K+ ATPase inhibition, consequent to this alteration of hyperpolarising/depolarising neurotransmitter ratio, can lead to neuronal degeneration.35 Reduced dopamine levels have been noticed in our study but there is increased dopaminergic transmission in HD. There is a possibility that circulating plasma levels of dopamine may not correlate with CNS dopamine levels. Also, increased levels of nicotine can lead on to dopamine release, promoting dopaminergic transmission even in the presence of reduced dopamine synthesis.36 The increase in serotonin levels and decrease in dopamine and noradrenaline could contribute to the psychiatric manifestations and cognitive dysfunction described in trisomy 21 and HD. Nicotine acts as a CNS stimulant and can bind to the central nicotinic receptors, contributing to the increase in cholinergic transmission and tremor in HD.36 Membrane Na+-K+ ATPase inhibition can lead on to increase glutamatergic excitatory transmission contributing to trisomy 21 and HD. In the presence of hypomagnesemia, consequent to membrane Na+-K+ ATPase inhibition, the magnesium block on the NMDA receptor is removed leading to NMDA excitotoxicity.37 The increased presynaptic neuronal calcium can produce cyclic AMP dependent phosphorylation of synapsins resulting in increased glutamate release into the synaptic junction and vesicular recycling. Increased intracellular calcium in the post synaptic neuron can also activate the calcium dependent NMDA signal transduction. The plasma membrane glutamate transporter (on the surface of the glial cell and presynaptic neuron) is coupled to sodium gradient which is disrupted by the inhibition of membrane Na+-K+ ATPase, resulting in decreased clearance of glutamate by presynaptic and glial uptake at the end of synaptic transmission. By these mechanisms, inhibition of membrane Na+-K+ATPase can promote excitatory glutamatergic transmission.37 Serotonin and quinolinic acid are NMDA agonist and positive modulators, and could contribute to increased NMDA transmission.38 Strychnine, by blocking glycinergic transmission, contributes to the decreased inhibitory transmission in the brain. Strychnine displaces glycine from its binding sites and the glycine is free to bind to the strychnine insensitive site of the NMDA receptor and promote excitatory NMDA transmission.36 Upregulated NMDA transmission could lead to increased excitatory transmission in the corticostriatal glutamatergic pathways and also produce derangement of the basal ganglia functional loops contributing to choreiform movements in HD. NMDA excitotoxicity contributes to neuronal degeneration in trisomy 21 and HD by increasing the intracellular calcium levels.39 All these results agrees with previous work on tryptophan catabolic pathways in Huntington's disease.6 The membrane Na+-K+ ATPase inhibition related magnesium depletion and elevated dolichol levels can upregulate the metabolism of glycosaminoglycans, glycoproteins and glycolipids.40 The results show an increase in the concentration of serum total GAG, individual GAG fractions, glycolipids and carbohydrate components of glycoproteins in trisomy 21 and HD. The increase in the carbohydrate components (total hexose, fucose and sialic acid) in trisomy 21 and HD was not to the same extent suggesting qualitative change in glycoprotein structure. The activity of GAG degrading enzymes and glycohydrolases was increased in trisomy 21 and HD suggesting reduced lysosomal stability and leakage of the lysosomal enzymes into the serum. Intracellular magnesium deficiency also results in defective ubiquitin dependent proteolytic processing of glycoconjugates as it requires magnesium for its function.41 Defective ubiquitin dependent proteolytic processing of proteins has been described in neuronal degeneration.42 The increase in the concentration of carbohydrate components of glycoproteins inspite of increased activity of many glycohydrolases may be due to their possible resistance to cleavage by glycohydrolases consequent to qualitative change in their structure. Structurally abnormal glycoproteins resist catabolism by lysosomal enzymes and accumulate in neuronal degeneration as in the case of beta-amyloid in trisomy 21.1 Proteoglycan complexes formed in the presence of altered calcium/magnesium ratios intracellularly may be structurally abnormal and resistant to lysosomal enzymes and may accumulate. Interaction between HS-proteoglycan and ChSproteoglycan with proteins like beta amyloid and huntingtin and reduced proteolytic digestion of these complexes can also lead on to their accumulation in the neurons.43 The upregulation of isoprenoid pathway can lead to increased cholesterol synthesis and magnesium deficiency can inhibit phospholipid synthesis. The cholesterol : phospholipid ratio of the RBC membrane was increased in trisomy 21 and HD. The concentration of total GAG and hexose and fucose of glycoprotein decreased in the RBC membrane and increased in the serum suggesting their reduced incorporation into the membrane and defective membrane formation. This could be due to inhibition of membrane trafficking enzymes (GTPases and lipid kinases) which transport membrane components from the endoplasmic reticulum - golgi complex, where they are synthesised to the cell membranes in the presence of magnesium deficiency.44 The change in membrane structure produced by alteration in glycoconjugates and cholesterol : phospholipid ratio can produce changes in the conformation of Na+-K+ ATPase resulting in further membrane Na+-K+ ATPase inhibition. The same changes can affect the structure of organalle membrane and result in defective lysosomal stability. The concentration of ubiquinone decreased significantly in trisomy 21 and HD which may be the result of low tyrosine levels, consequent to digoxin's effect in preferentially promoting tryptophan transport over tyrosine.5 The aromatic ring portion of ubiquinone is derived from the tyrosine. Ubiquinone, which is an important component of the mitochondrial electron transport chain, is a membrane antioxidant and contributes to free radical scavenging. The increase in intracellular calcium can open the mitochondrial PT pore causing a collapse of the hydrogen gradient across the inner membrane and uncoupling of the respiratory chain.45 Intracellular magnesium deficiency can lead to a defect in the function of ATP synthase. All this leads to a defect in mitochondrial oxidative phosphorylation, incomplete reduction of oxygen and generation of superoxide ion which produces lipid peroxidation. Ubiquinone deficiency also leads to reduced free radical scavenging. The increase in intracellular calcium may lead to increased generation of NO by inducing the enzyme nitric oxide synthase which combines with superoxide radical to form peroxynitrite. Increased intracellular calcium also can activate phospholipase A2 resulting in increased generation of arachidonic acid which can undergo increased lipid peroxidation. Increased generation of free radicals like the superoxide ion and hydroxyl radical can produce lipid peroxidation and cell membrane damage which can further inactivate membrane Na+-K+ ATPase triggering the cycle of free radical generation again. There was an increase in lipid peroxidation products and NO with decreased antioxidant protection as indicated by a decrease in ubiquinone and reduced glutathione levels and free radical scavenging enzyme activity in trisomy 21 and HD. Glutathione synthetase, glutathione peroxidase and glutathione reductase are dysfunctional in the presence of magnesium deficiency. The increased intracellular calcium related opening of the mitochondrial PT pore produces hyperosmolality and matrix expansion of the mitochondria. This ruptures the outer membrane producing leakage and inactivation of the mitochondrial dismutase. Alteration in peroxisomal membranes results in a catalase dysfunction. Mitochondrial dysfunction related free radical generation has been implicated in the pathogenesis of neuronal degeneration like HD and trisomy 21. Mitochondrial dysfunction can remove the magnesium block of the NMDA receptor leading on to excitotoxicity and neuronal degeneration.46 Cell death is also mediated by the increased intracellular calcium and ceramide related opening of the mitochondrial PT pore causing a collapse of the hydrogen gradient across the inner membrane. This leads to volume dysregulation of mitochondria causing hyperosmolality of matrix and expansion of matrix space. The outer membrane of the mitochondria ruptures and releases AIF (apoptosis inducing factor) and cyto C (cytochrome C) in to the cytoplasm activating caspase-9, important in apoptosis crucial to neuronal degeneration. Also caspase 3 activation can cleave P21 involved in linking DNA duplication to cell division resulting in a polyploid cell and oncogenesis noticed in trisomy 21. The isoprenoid pathway may this contribute to the genesis of trisomy 21 and Huntington's disease, the two neurogenetic syndromes. Thus, trisomy 21 and HD are evidence of the role played by the central nervous system in regulating genomic function. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02048t4.jpg] [ni02048t1.jpg] [ni02048t3.jpg] [ni02048t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}