 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 50, No. 3, Sept, 2002, pp. 337-339 Case Report Atypical Benign Partial Epilepsy of Childhood (Pseudo-Lennox Syndrome) : Report of Two Brothers G. Deda, H. Caksen Department of Pediatrics,
Division of Pediatric Neurology, Ankara University,
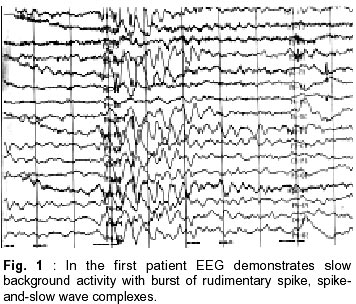
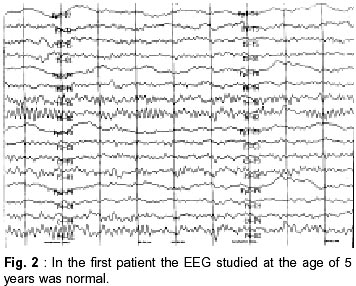
Faculty of Medicine, Ankara, Turkey. Accepted for publication : 6th November, 2000. Code Number: ni02096 Summary Two brothers (3 and 2 year old) with characteristic findings of atypical benign partial epilepsy of childhood (pseudo-Lennox syndrome) are reported, to emphasize the presence of a possibility of a genetic basis of this disorder and the importance of intravenous immune globulin (IVIG), vigabatrin (VGB) and lamotrigine (LTG) therapy. Sleep EEGs of both the patients showed typical features of Lennox-Gastaut syndrome. On follow-up, the convulsions were found to be resistant to numerous antiepileptic agents in one patient while they were easily controlled with LTG monotherapy in the other patient. In the elder brother, who was diagnosed as intractable epilepsy, the convulsions disappeared with IVIG and VGB. During the long term follow-up, they were seizure free for five and two years respectively, and their mental motor development was excellent. Key words : Atypical partial benign epilepsy, Immune globulin, Vigabatrin, Lamotrigine. Introduction In 1982, Aicardi and Chevrie1 reported seven children with an unusual epileptic syndrome and named the condition 'atypical benign partial epilepsy of childhood' (ABPE). The main clinical features include onset between 2.5 years and six years of age; the occurrence of several types of seizures, especially partial motor seizures, atypical absences and myoclonic-atonic seizures; and the persistence of normal neurological and mental function throughout the course.1 Some authors have used the term 'pseudo-Lennox syndrome' for this epileptic syndrome.2,3 The authors report two brothers with typical findings of ABPE to emphasize the presence of a possibility of genetic cause of this disorder and the importance of intravenous immune globulin (IVIG), vigabatrin (VGB) and lamotrigine (LTG) therapy in ABPE. Case Reports Case 1 : A 3 year old boy was admitted with history of convulsions. The pregnancy, labour and delivery were unremarkable, but he had an exchange transfusion due to indirect hyperbilirubinemia in the neonatol period. His developmental milestones were normal. He was the first child of non-consanguineous parents. His father's cousins had epilepsy since childhood. The physical examination was normal. On laboratory investigation, routine urine and blood analyses, liver and renal function tests, ammonia, lactic and pyruvic acid levels, urine and blood amino acids were normal. Computerized tomography of the brain was normal. He had first generalized tonicclonic convulsion, with high fever at the age of two years and later (at the age of two years and 10 months) two afebrile seizures occurred. At that time, phenobarbital (PB) therapy was initiated in another hospital. Inspite of PB, the generalized tonic-clonic convulsions continued about nine times in few days. PB was replaced by valproic acid (VPA) as EEG had revealed generalized paroxysmal sharp waves. After initiation of VPA therapy, the generalized tonic-clonic convulsions occurred approximately 10-15 times a day. Thereafter, VPA therapy was discontinued and the treatment with phenytoin (PHT) and clonazepam (CLN) were initiated in another hospital. Despite PHT and CLN therapy, the seizures occurred 30-40 times a week and lasted 1-2 minutes. The patient was hospitalized with the diagnosis of intractable epilepsy in order to observe the type of convulsion and management. Different combinations of PHT, carbamazepin (CBZ) (20 mg/kg/day), VPA (30 mg/kg/day) and CLN (0.1 mg/kg/day), allopurinol (13 mg/kg/day), primidone (7 mg/kg/day)) and barbexaclone (5 mg/kg/day) were tried without any benefit. However the seizures, mainly generalized tonic-clonic but sometimes absence and atonic episodes continued 3-9 times a day. EEG showed slow background activity with burst of rudimentary spike, spike-and-slow wave complexes. These findings of EEG were compatible with Lennox-Gastaut syndrome (LGS) (Fig. 1). The seizures were still not controlled. On the 30th day of admission, IVIG (400 mg/kg/day) was administered. The seizures decreased in frequency and subsequently, completely disappeared after three days of IVIG therapy (IVIG therapy was given for five days). The patient was discharged from hospital on primidon, barbexaclone and VPA (35 mg/kg/day). One month after discharge from the hospital, he started having generalized tonic-clonic convulsions, about five times a day. Primidon and barbexaclone were stopped and vigabatrin (VGB) was added. After initiation of VGB therapy, the seizures ceased. The EEG studied at the age of 5 years was found to be normal (Fig. 2). However, it demonstrated active epileptiform anomaly on the right hemisphere at the age of 8 years. The combination of VGB and VPA was used for four years. He was followed on treatment with VPA, and was seizure-free for five years. At the age of 9 years, his mental and motor development was remarkable with normal scholastic performance. Case 2 :A 2.5 year old boy (the younger brother of the first case) was admitted with history of convulsions. He was born preterm and was managed with the diagnosis of wet-lung in the newborn unit for three days. At that time, he had prolonged indirect hyperbilirubinemia, however all the tests were normal. His developmental milestones, physical examination and routine urine and blood analyses, liver and renal function tests, urine and blood amino acid test were normal. MRI of the brain was also normal. The patient's first seizure episode occurred 10 days before admission to the hospital. The attacks were characterized by myoclonic-atonic seizures, and absence episodes, which occurred 7-8 times/day. They lasted a few seconds. EEG showed burst of rudimentary spike and slow wave complexes. These findings of EEG were compatible with LGS. The patient was hospitalized and lamotrigine LTG (1 mg/kg/day) was initiated. On the first day of admission, only one seizure was observed. He was discharged from hospital on the 7th day. During the 8th and 13th month of follow-up, he had two similar type of convulsions with high fever. The LTG dose was increased to 2.5 mg/kg/day. On follow-up at 8th month, EEG showed spike slow wave complexes at about 2.5-3 Hz. At the age of 6 years, he was seizurefree for two years and his mental-motor development was excellent. Discussion The exact nosological situation of ABPE is not completely clear, especially in relation to the so called electrical status epilepticus of slow sleep.4 Nieto- Barrera et al5 suggested that the continuous spikewave during slow sleep syndrome (CSWS), Landau- Kleffner syndrome (LKS) and ABPE have probably the same pathophysiology; and that they are the severe, moderate and benign forms of a single epileptic syndrome, age-related, with continuous spike waves and various neuropsychological and behavioral disturbances. Yoshimura et al6 reported identical twins with ABPE and stated that a possibility of genetic cause may be there in this disorder.We also suspect a possibility of genetic cause in ASPE, as it was diagnosed in two brothers in our study. In addition, the presence of epilepsy in cousin of the patient's father also supports the possibility of genetic cause in ABPE. Exact nature of seizures, however, cannot be commented upon in these relatives as they were not examined by the authors. In ABPE, several types of seizure may be observed e.g. partial motor seizures, atypical absences, atonic and/or myoclonic seizures, and generalized tonicclonic seizures. Atonic and/or myoclonic seizures occur often in clusters and recur many times daily. Atonic attacks are particularly prominent and may occur several dozen times a day, often with falls. The clusters may last two to three weeks and be separated by periods of months.1,7,8 We also diagnosed several type of seizures in our patients, including generalized tonic-clonic, absences, atonic and myoclonic seizures. The sleep EEG of such children shows continuous spike waves during slow sleep. The awake EEG shows multiple bilateral discharges of spike and wave complex.1,7,9 Such patients often receive an erroneous diagnosis of LGS, although the course of this epileptic syndrome is generally favourable, with spontaneous remission before the age of 10 years.1,2 In accordance with the literature, of both the cases reported here, sleep EEG showed typical features of LGS. They were also initially diagnosed as LGS; however during the their follow-up LGS was ruled out and they were diagnosed as ABPE. It has been reported that the seizures in this condition are resistant to treatment with several antiepileptic drugs.8 In various studies, VPA, CLN, PHT, sulthiame, sulthiame/clobazam, adrenocorticotropic hormone and ketogenic diet have been successfully used in the treatment of children with ABPE.6,8,10,11 Gross-Selbeck10 noted that CBZ used in ABPE usually has no effect, either on the seizures or on the EEG. On the contrary, in some cases both may even get worse. In the present study, particularly in the first case, numerous anticonvulsants were used, but the seizures were not controlled. During the follow-up, dramatic improvement was achieved with the treatment of IVIG and subsequently VGB. In the second case, the seizures were easily controlled with only LTG, which was not used in the first patient. In the first case, only one course of IVIG (the dosage of 400 mg/kg/day for five days) was used because the protocol of IVIG therapy was not completely determined in our clinic at that time. In conclusion, a possibility of genetic basis does exist in ABPE and that LTG,VGB or IVIG may be preferred as the initial treatment of choice. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02096f1.jpg] [ni02096f2.jpg] |
| |||||||||

{kind=link}
{kind=link}