 |
search
for |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 50, No. 4, Dec, 2002, pp. 408-416 Review Article Multifocal Motor Neuropathy A. , M. Donaghy* Department of Neurology,
Derriford Hospital, Plymouth PL6 8DH
and
Department of Clinical Neurology*,
University of Oxford,
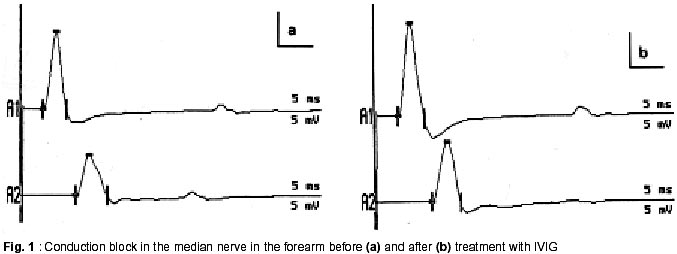
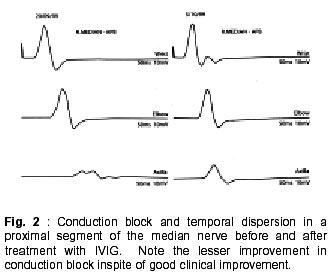
Radcliffe Infirmary, Oxford OX2 6HE, UK. Accepted for publication : 5th August, 2002. Code Number: ni02114 Summary Multifocal motor neuropathy (MMN) is a disorder characterized by the presence of chronic asymmetric, predominantly distal, lower motor neuron weakness commonly involving the upper limbs. Sensory involvement is minimal or absent. The presence of conduction block is a characteristic electrophysiological feature although other features like temporal dispersion may be equally relevant. An autoimmune basis has been suggested and antiganglioside antibodies like IgM anti-GM1 may be raised. Treatment with intravenous immune globulin is effective in most patients but repeated infusions are needed. Cyclophosphamide is useful but toxic. Steroids or plasma exchange are usually ineffective and may even be harmful. Other treatments like beta-interferon and rituximab have recently been used. It is important to recognize this treatable disorder from motor neurone disease and other lower motor neurone syndromes. Key words : Multifocal motor neuropathy, Conduction block, Temporal dispersion, Pathophysiology, Pathogenesis, Therapy, Intravenous immune globulin. Introduction The current interest in multifocal neuropathies with conduction block began in 1982 with the description of five patients with this syndrome.1 All five patients had chronic asymmetric sensorimotor neuropathy. A few years later patients with pure motor neuropathies with multifocal conduction block began to be described.2-4 Multifocal motor neuropathy (MMN) is now recognized as a relatively rare disorder characterized by slowly progressive, asymmetrical and predominantly distal lower motor neurone weakness with minimal or no sensory symptoms and with electrophysiological evidence of motor conduction block.5-8 Tentative estimates suggest the prevalence of MMN to be in the order of 1-2 per 100,000.8 Although parallels have been drawn with chronic inflammatory demyelinating neuropathy (CIDP) and motor neurone disease, MMN is now thought to be a separate clinical entity with distinct characteristics. In spite of this, misdiagnosis is common, even among neurologists, and may delay appropriate management by many years. In a Mayo Clinic study, only 6/46 patients with MMN were referred with the correct diagnosis.7 Recognition of this treatable disorder from motor neurone disease and other lower motor neurone syndromes is particularly important. Clinical Features Men are more frequently affected. Symptoms arise very insidiously in most patients, although subacute onset can also occur. The mean age at onset is around 40 years. Patients below 20 years of age and above 75 years of age at the time onset are infrequently reported. Asymmetric involvement at onset is seen in over 90% of patients and may persist throughout the illness. Weakness in upper limbs is much more common than in the lower limbs. Distal weakness at onset is found in at least 95% of patients with MMN, and in most patients tends to predominate through the illness. Weakness in the distribution of individual nerves may be recognized particularly in the early stages. The ulnar, median, radial, peroneal and tibial nerves are commonly affected. Finger drop or wrist drop are typical early signs reported by around onethird of the patients.6 Weakness of extension of an individual finger is a common early symptom. Fasciculations and cramps are common and have been variably reported in one-third to one-half of all patients. Fatigue can occur in the affected limbs. A worsening of the weakness following exposure to cold is interesting as it would not normally be expected to occur in demyelinating neuropathies. Muscle wasting may be mild or even absent in some patients, especially in the earlier years of illness. The relative absence of wasting in severely weak muscles indicates the presence of conduction block in the afferent nerve, as opposed to permanent axonal degeneration. On the other hand, early amyotrophy, sometimes within one year of onset, may characterize a separate group of MMN patients.6 This latter group has often been associated with poor recovery, attributed to irreversible axonal loss. Patients may simultaneously have severe wasting of some muscles, and profound weakness but minimal or no wasting of other muscles in the same limb. Of interest is a recent description of patients with an axonal type of MMN, some of who responded to immunomodulatory treatment.9 It is likely that the improvement in these patients, as well as those with typical MMN with conduction block reflects the improvement in the biophysical properties of the axons that are still viable. Tendon reflexes are frequently depressed, particularly in the affected limbs, but can be normal or even brisk. However spasticity, clonus or extensor plantar responses do not occur. Sensory symptoms, like parasthesiae, may be present in some patients but pain, other than cramp pain, is distinctly unusual. Sensory signs are usually absent or minimal. Autonomic involvement is unusual.7 Cranial nerve involvement can be seen very occasionally.5,10 Respiratory muscle weakness can rarely occur due to phrenic nerve palsy.11The long-term prognosis in patients with MMN is generally good, although some residual impairment is not uncommon, even in patients who respond to therapy. In the untreated or non-responsive patient, symptoms typically progress slowly, sometimes in a step-wise manner with years of stability in between, although spontaneous remissions can occur.6 Fatalities are rare. Pathophysiology of Conduction Block The presence of multifocal conduction block (CB) helps to differentiate MMN from other chronic lower motor neurone syndromes. Conduction block occurs when an action potential fails to propagate through a stretch of structurally intact axon. It is measured as the distal to proximal reduction in area or amplitude of the compound muscle action potential (CMAP) and is traditionally expressed as a percentage. Normal conduction through a myelinated axon is efficient and speedy and is enabled by the process of saltation. Inward action currents through sodium channels at the activated node of Ranvier generate outward driving current at the next node to be excited. This in turn depolarizes the node to threshold, opening up sodium channels and the cycle is repeated. For successful conduction to take place the driving current must exceed the threshold current at the node by a ratio known as the safety factor. In normally myelinated axons the safety factor is usually greater than 5. In demyelination, some of the current leaks out through the paranodal regions due to increased capacitance and decreased resistance of the membrane. The driving current takes longer to charge the next nodal membrane, the internodal conduction time is prolonged and conduction is slowed. If the driving current becomes insufficient to overcome the threshold current, the safety factor falls below 1 and conduction block occurs. Additional contribution to conduction failure may come from the activation of the paranodal fast potassium channels.12 These fast channels remain inactive during normal saltatory conduction. In the demyelinated fibre, current dissipation through the paranode depolarizes the paranodal axon, which in turn activates the flow of outward repolarizing currents through the exposed fast potassium channels. The already weakened driving current is further compromised and may precipitate conduction block. It is common to see patients who complain of muscle fatigue and weakness in areas where conduction block either cannot be found or has remained unchanged during conventional neurophysiological examination. These symptoms could be explained by the occurrence of activity-dependent conduction block.13 During muscle activity, a train of impulses conducted through the axon produces a rise in the axoplasmic sodium content. The rise is higher in demyelinated axons to make up for current dissipation. The excess sodium triggers the electrogenic sodium-potassium pump, which pushes out more sodium than it takes in potassium. The membrane hyperpolarizes and when the threshold is elevated beyond the already compromised driving current, transient conduction block occurs. Recent evidence suggests that hyperpolarizing block may be part of the story. It does not explain all symptoms that are seen in MMN patients, for example, the occurrence of cold paralysis. Cold paralysis has been reported in patients with monomelic amyotrophy,14 but would be unusual for most demyelinating states. The primary biophysical defect in MMN may be the coexistence of membrane hyperpolarization juxtaposed to focal areas of depolarization.15 Focal damage to the axon, by edema or by immune-mediated mechanisms for example, could block the electrogenic sodium-potassium pump activity. This would create a depolarization block at the site, and an increase in intra-axoplasmic sodium content. Accumulation of extracellular potassium, possibly due to breakdown of the blood-nerve barrier, could also contribute. The excess sodium could diffuse longitudinally along the axon to the adjacent sites where the pump is still working, thereby producing membrane hyperpolarization in those areas. Continuing sodium influx would eventually lead to a steady state of sodium flow between the depolarized and hyperpolarized segments. Thus depolarization at the site of the lesion would coexist with chronic hyperpolarization, due to persistent activity of the sodium-potassium pump, on one or both sides of this site. The hyperpolarized segment, behaving like a post-ischemic nerve segment, could generate ectopic activity that is clinically manifested as fasciculations or myokimia.12,16 Cold paralysis could be due to the increase in the depolarizing conduction block caused by slowing down of the temperature-sensitive sodiumpotassium pump at lower temperatures. Axonal degeneration, frequently seen in MMN, could be due to excessive intracellular calcium ions caused by a reversal of the sodium-calcium exchange mechanism in the persistently depolarized segment.15 In MMN, conduction block is seen along motor, but not sensory nerves, and at sites other than common areas of entrapment. The peculiar sparing of conduction through sensory fibres is interesting and may be due to a number of different biophysical properties between human motor and cutaneous afferent axons. Some of these properties may tend to protect sensory axons from excessive hyperpolarisation. The same pathology would then make the motor nerves more susceptible to conduction failure compared to cutaneous afferents. Diagnosis of Conduction Block in MMN Conduction block is measured as the distal to proximal reduction in area or amplitude of the compound muscle action potential (CMAP) and is expressed as a percentage. Various working definitions of CB have been proposed, with values varying between 20% and greater than 50%.7,17-24 Many definitions assess changes in the negative peak values, although peak-to-peak measurements have also been used. A major obstacle to the satisfactory definition of CB has been the presence of abnormal temporal dispersion (TD), caused by non-uniform conduction across a nerve segment. Non-uniform conduction velocities not only prolong the duration but also produce desynchronisation of the CMAPs in individual nerve fibres, which in turn leads to the cancellation of the positive peaks with some of their negative peaks. The proximal CMAP amplitude or area drops as a result of this 'interphase cancellation' and can produce a pattern of pseudo-conduction block.18 This can be further exaggerated in chronic axonal loss, where the CMAPs are of small amplitude and frequently polyphasic. Because of these reasons, many of the existing definitions of CB either do not accommodate TD or demand even more stringent criteria in its presence. 7,17-19,22 This could reduce sensitivity for electrophysiological detection of this potentially treatable disorder. The relation between TD and drop in CMAP amplitude or area is likely to be more complex. Temporal dispersion and conduction block are known to co-exist in the same nerve segment (Fig. 1).25,26 Our studies, as well as those by others, suggest that temporal dispersion could be important and independent part of the electrophysiology of MMN.21,25 The explanation may lie, among other factors, with the relative balance between delay and block of axonal conduction among the faster and the slower conducting fibres. Thus, disproportionate slowing through the slower conducting fibres could prolong the duration of the CMAP, but produce little change in amplitude. Block in conduction mainly affecting slower fibres as opposed to the faster conducting ones could produce a lesser reduction in CMAP amplitude and a shorter CMAP duration. On the other hand, a non-uniform slowing of conduction (without actual block of axonal conduction) through the faster conducting fibres could lead to increased interphase cancellation and pseudo-conduction block with or without TD,18 while complete block through these same fibres would produce true CB. Slowing or blocking of conduction through the fastest conducting fibres could, in addition, slow the conduction velocity across that segment or prolong the distal latencies. In reality both non-uniform slowing and true conduction block are likely to coexist in varying proportions. Quantification of conduction block may, therefore, be difficult and would depend upon the subpopulations of axons that are predominantly affected.18 Evolution from minor to major block could occur over time,20 suggesting dynamic changes in axonal conduction. In keeping with the clinical findings, conduction block is more often detected in the arms than in the legs. Electrophysiological and radiological evidence suggests that at least one-third of conduction blocks may be found proximally. Special techniques for cervical stimulation may have to be employed for selected patients. In more accessible regions like the forearm, the exact site of conduction block can sometimes be accurately localized by short segment stimulations or 'inching'.27 An abrupt drop in the CMAP amplitude indicates focal conduction block as opposed to the gradual drop as can be seen in chronic demyelinating neuropathy or after chronic axonal loss. A few patients with MMN may not have detectable conduction block inspite of an adequate search. Arguably, some of the conduction block may be very proximal and at effectively inaccessible sites. More commonly, relatively stringent criteria for conduction block might have been used. There could also be axonal variant of MMN without conduction block or other features of demyelination.9 Other features of demyelination commonly seen in MMN patients include prolonged distal motor latencies and reduced conduction velocities especially across the affected segments. Prolonged or absent Fwaves may indicate proximal conduction block. Needle electrode recordings may be indistinguishable form those seen with anterior horn cell involvement or in motor axonopathies and include fibrillations and positive sharp waves, fasciculations, large or polyphasic motor unit potentials (MUPs) and reduced recruitment. Indeed, reduced MUP recruitment out of proportion to the change in MUP morphology should make one suspicious of conduction block, in the appropriate clinical context. Further Investigations IgM anti-GM1 and other antiganglioside antibodies including those against asialo-GM1, GD1a and GM2 can be variably elevated in 20% to 85% of patients with MMN using ELISA techniques.28,29 The success in detection may depend on the technique used for assay. The presence of IgM anti-GM1, although helpful, is neither essential nor specific for the diagnosis of MMN. Anti-GM1 antibodies may be raised to lesser extent in some patients with other lower motor neurone syndromes, CIDP and in Guillain Barre syndrome, where they are typically of the IgG variety. Also, there is no difference in the clinical or electrophysiological features between patients with or without elevated anti-GM1 levels.7 However, in a patient with a pure lower motor neurone syndrome, a raised IgM anti-GM1 level may be strongly predictive of MMN. Recently, biosensor assay techniques are being developed that offer a rapid system for measuring anti GM1 antibody levels, while retaining sensitivity and specificity that are comparable to those found with the ELISA system.30 Serum creatine kinase levels may be mildly elevated. A monoclonal paraprotein can be found in a minority of patients. Polyclonal increase in immunoglobulins is seen in some patients. MMN may coexist with other myeloproliferative or lymphoproliferative disorders, mostly of the B-cell type. In our centre, we have seen patients with MMN who were also being treated for IgM MGUS, Waldenström's macroglobulinaemia, essential thrombocythaemia, Hodgkin's lymphoma or malt lymphoma of the conjunctiva. Cerebrospinal fluid protein levels, if raised, are rarely above 1g/l. Oligoclonal bands are not seen. MR scans of the brachial plexus may show asymmetric high signal in T2-weighted images.31 This is in contrast to symmetrical distribution of similar T2-weighted signal changes in CIDP patients and normal signals in MND. Consecutive slice T1WI of the median nerve in the forearm have shown focal gadolinium enhancement at sites that correspond to areas of conduction block.32 Pathology and Immunopathogenesis Given the predominantly motor involvement in MMN, histopathological studies of nerves have been infrequent. Where this has been done, motor or mixed nerves tended to show demyelination, sometimes with onion bulb formation, or axonal degeneration.32-35 Pathological changes could sometimes be localized to the site of the conduction block.32 The majority of the biopsies of the sural or other sensory nerves have either been normal or have shown mild loss of axons, or demyelination, or both.5,6,33,36 The pathogenesis of MMN is obscure. Clinical improvement with immunoglobulins suggests that there might be an underlying immune-mediated mechanism. Some researchers propose a pathogenic role for anti-GM1 antibodies but this remains debatable. Anti-GM1 levels may be raised in MMN. In some patients the levels could be reduced by successful treatment.37 The GM1 ganglioside is distributed widely throughout the nervous system. Antibodies from patients have been shown to bind to nerve roots, nodes of Ranvier, and intramuscular motor nerve endings suggesting a potential role for these antibodies. Immunisation of rabbits with anti- GM1 or Gal (beta 1-3) GalNAc produced an antibody response, conduction block, axonal degeneration and immunoglobulin deposition at the nodes of Ranvier.38 When serum of a patient with a motor syndrome, conduction block and high titres of anti-GM1 was injected along with fresh complement into a rat sciatic nerve, it induced conduction block, temporal dispersion, immunoglobulin deposition at the node of Ranvier and mild demyelination. Pre-absorption of the patient's serum with GM1 removed the binding activity and the ability to induce conduction block, suggesting that anti-GM1 antibody was responsible for the damage.39 Others have also produced conduction block by injecting sera from patients with high anti-GM1 titres into rat nerves.40 Takigawa et al suggested that anti-GM1 antibodies, in the presence of complement could block nodal sodium channels.41 This led to the hypothesis that sodium channel blockage by the antibody may play a pathogenic role in some motor neuropathies like MMN and axonal Guillain Barre Syndrome.42 43 On the other hand, others have failed to show significant conduction abnormalities caused by application of anti-GM1 fractions,44-46 sometimes in spite of binding of anti-GM1 to the nodes of Ranvier.44,46 The sodium-channel blocking effect of anti-GM1 sera has also been questioned.45 Indeed, there have been unproven suggestions that sodium influx through channels may actually be increased at the site of the lesion.15 Neither the diagnosis of MMN nor its improvement to treatment seems to depend on the presence or absence of anti-GM1 antibodies in the serum of patients.5,6,21,24,33,47 At least for the time being, the relative role of antiGM1 antibody in the pathogenesis of MMN remains to be confirmed. Treatment The suspicion that MMN might be immune-mediated has led to the search for treatment with immunomodulating therapy. However, steroids, given orally or intravenously in high doses are unusually ineffective and often induce deterioration.5,10,33,37,48 Plasma exchange is usually ineffective.10,37,49 Immunoadsorption and CSF filtration have not helped.35,50 The first reported benefits came with the use of intravenous cyclophosphamide (CTX).37,51 In those who respond, the benefits may take two to five months to appear.37 Response to CTX may accompanied by a progressive reduction in the anti-GM1 levels. Discontinuation of this treatment may not only lead to clinical relapse but in some patients, with a rise in antibody levels. Long-term use of high dose CTX is potentially toxic, making this form of therapy less suitable for the young, for those with less severe disease and for those who may respond to other, less toxic regimes. Oral cyclophosphamide, used alone, has only rarely been beneficial.10 On the other hand, low-dose oral cyclophosphamide added to patients responsive to intravenous immunoglobulin (IVIG) may reduce the frequency with which IVIG is required and may even help to stop the IVIG infusions altogether.33,52,53 Cyclophosphamide remains an option in patients with substantial disability due to conduction block, but who fail to respond to IVIG. The place of high-dose IVIG in the treatment of MMN has now been established.5,10,33,54,55 Almost 80% of MMN patients respond to IVIG therapy.8 The usual recommended dose is 2g/kg given as an infusion over 2 to 5 days. The treatment is repeated in cycles depending upon the duration of response. Most patients who respond to IVIG tend to do so from the first cycle, although some incremental benefit through the first few cycles may be seen. Clinical improvement is usually prompt and may occur within the first 48 hours. Restoration of functional abilities is frequently the first and most obvious response. The improvement tends to build up over one or two weeks and usually lasts for several more weeks before wearing off. In some patients, the initial duration of benefit regresses, but increasing the frequency of the cycles of IVIG tends to restore stability. Depending upon the response, individual cycles of IVIG may be needed every one to eight weeks in the majority of patients. Most patients will need long-term therapy. A minority may show a cumulative benefit over several cycles, lessening the need for IVIG, and in a few instances leading to permanent remission.52 In patients requiring long term repeated infusions hometherapy is useful so as to avoid hospital attendance and to maintain strength on an optimal plateau, avoiding treatment-related fluctuations . The pattern of response to IVIG is typically stereotyped. Recently affected muscles and those with minimal or no wasting show the best improvements. Conduction block or temporal dispersion may variably improve.10,19,25,33,56 IVIG has little effect on the anti-GM1 levels. How IVIG acts in MMN remains unclear. Proposed mechanisms of action include supply of idiotypic antibodies, neutralization of pathogenic cytokines, inhibition of complement binding, down-regulation of antibody production, modulation of Fc-receptor function, modulation of Tcell function, and enhancement of remyelination.57 IVIG remains the most effective and a relatively safe treatment for MMN, but the prohibitive long-term costs raises serious logistic issues. Adding low-dose oral CTX to the regime may help a selected group of patients but not others.33,52,53 A few patients with MMN will not respond to IVIG at all. Early evidence suggests that some of them may benefit from interferon b1a therapy.58 Improvement due to interferon b1a tends to start after a few weeks of therapy and last longer than the effect of IVIG. Azathioprine can stabilize a few IVIG unresponsive patients.6 Rituximab, a monoclonal antibody directed against B-cells has been used successfully in a few patients.59 Both clinical improvement and a lowering of anti-GM1 levels were seen. Differential diagnosis The place of MMN in the context of other dysimmune neuropathies is still debated. It is not difficult to separate the asymmetric, distal, motor pattern of MMN from typical CIDP. Typical CIDP is characterized by a chronic, symmetrical, sensorimotor neuropathy with generalized hypo- or areflexia, raised CSF protein levels and obvious electrophysiological features of demyelination, even in clinically unaffected areas.60 CIDP responds well to steroids or plasma exchange as well as to other immunomodulating therapy. Even the pure motor variants of CIDP tend to be symmetrical, although sometimes predominantly with hand involvement.61A more intermediate pattern may have features similar to both CIDP and MMN, and has been variably called it multifocal demyelinating neuropathy, multifocal inflammatory demyelinating neuropathy, multifocal motor and sensory demyelinating neuropathy or Lewis-Sumner syndrome, or simply a variant of CIDP. Others see it as a part of the clinical spectrum of MMN. Like MMN, multifocal demyelinating neuropathy is asymmetric and tends to start in the arms. However sensory symptoms and signs may be prominent. Like MMN, the CSF protein levels are often normal or mildly raised and MR scans of the brachial plexus may show asymmetric signal changes. Patients with multifocal demyelinating neuropathy may respond to steroids unlike MMN patients. Recently 9 patients with a pure axonal variant of MMN have been described.9 Their clinical features were similar to that seen in typical MMN, but no conduction block or other features of demyelination were detected and the anti GM1 antibodies were not raised. Three patients responded to IVIG treatment. Although the authors report it as a distinct clinical entity from MMN, at least some of the features described in a few patients suggested the presence of electrophysiologically inaccessible conduction block.62 Patients with acute monophasic motor neuropathy with predominant upper limb involvement and raised anti-GM1 antibodies have been described.63,64 In some cases this followed a recent Campylobacter jejuni enteritis. The monophasic pattern of the illness with progression for four to five weeks followed by spontaneous recovery and the presence of raised CSF protein levels are suggestive of a variant of acute polyneuritis rather than MMN. It may be difficult to distinguish MMN from a pure lower motor neurone variant of MND.3,4 Asymmetric onset in the upper limb with slow progression can occur in MND and a few patients may also have elevated anti GM1 antibody levels. Fasciculations and cramps are common to both disorders. Retained or brisk reflexes in wasted muscles in a patient with MMN may cause further confusion. As discussed earlier, chronic axonal loss may produce a false appearance of conduction block. Needle electrode recordings of muscles are indistinguishable, but may be more extensive in MND, and can be helpful especially in the earlier stages of presentation. Amongst the asymmetric lower motor neurone syndromes, monomelic amyotrophy (Hirayama disease)14,65 may be particularly difficult to differentiate from MMN on clinical grounds. It is a benign, sporadic lower motor neurone syndrome of young adulthood typically presenting with distal unilateral weakness and atrophy, most commonly in the hand and forearm. Males are predominantly affected. Spontaneous arrest after a few years of slow progression is common. Like other anterior horn cell disorders, wasting is early and can be quite pronounced. Tendon reflexes are usually depressed or absent, but may be retained, or rarely brisk.65 However, other upper motor neurone signs do not develop. Central motor conduction studies in patients with Hirayama disease failed to show evidence of pyramidal dysfunction.66 Cold paralysis is a curious symptom that is common to both MMN and monomelic amyotrophy.14,65 A few patients with monomelic amyotrophy who had proximal conduction block have been reported.67 Although monomelic amyotrophy has traditionally been seen as a variant of motor neurone disease, recent MR scan studies have shown focal atrophy of the lower cervical cord and anterior unfolding of the dural sac in neck flexion.68-70 This in contrast to the asymmetric high signal changes in the brachial plexus that may be seen in MMN. Neurogenic thoracic outlet syndrome can present with unilateral atrophy and weakness of hand muscles and sensory signs may be minimal or absent. Pain may be prominent and typical electrophysiological findings include reduced amplitudes of ulnar sensory and medial motor responses. Sensory action potentials from the medial antebrachial cutaneous nerve may be reduced. Future directions Our understanding of MMN has improved steadily since it was first described 17 years ago. However, there is still a long way to go. The prevalence of MMN is still uncertain, although this might change with better recognition in future. The full clinical spectrum of MMN and its relation with CIDP or MND has not been fully clarified. More pragmatic definition of conduction block is necessary for diagnosis, and prediction of IVIG responsiveness, and for objective measurement of treatment responsiveness. Our interpretation of the pathophysiology remains rudimentary despite improving understanding of axonal conduction. The role, if any, of antiganglioside or other antibodies in the pathogenesis of MMN is not yet clear. Newer treatments need to be developed, as the current options are either too expensive or relatively toxic. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02114f2.jpg] [ni02114f1.jpg] |
| |||||||||

{kind=link}
{kind=link}