 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 50, No. 4, Dec, 2002, pp. 444-451 Glioneuronal Migration and Development Disorders : Histological and Immunohistochemical Study with a Comment on Evolution L. Pal, S.K. Shankar,* V. Santosh,* T.C.Yasha* Department of Pathology,
Sanjay Gandhi Postgaduate Institute of Medical Sciences, Lucknow
and
Department of Neuropathology*,
National Institute of Mental Health and Neurosciences,
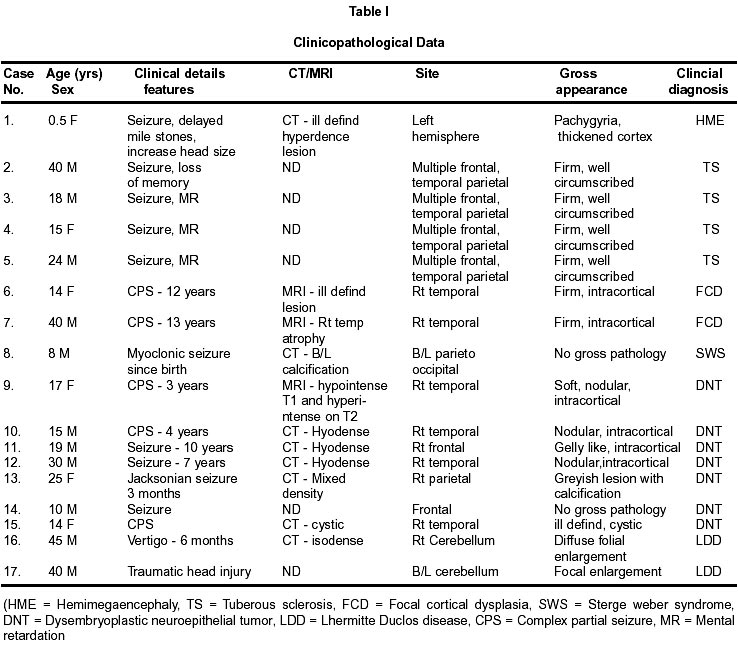
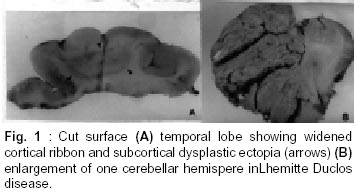
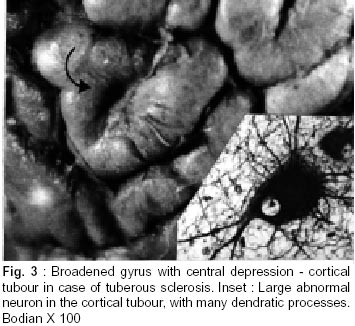
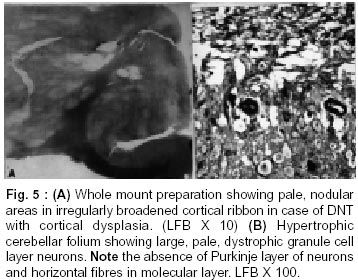
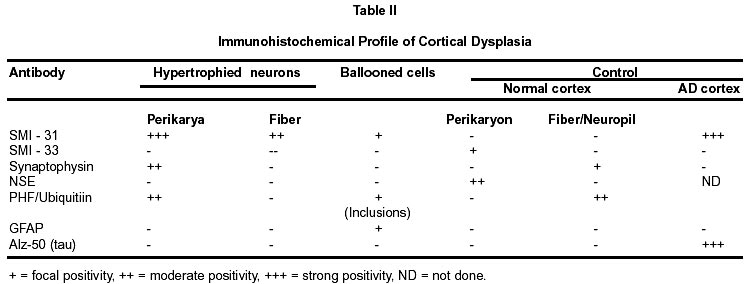
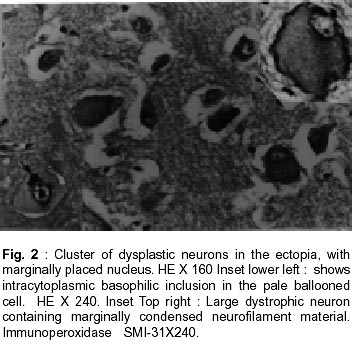
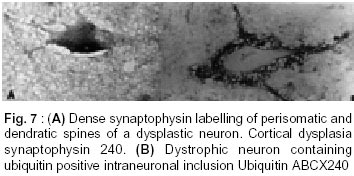
Bangalore - 560 029, India. Accepted for publication : 2nd August, 2001. Code Number: ni20120 Summary Glioneuronal migration disorders of the brain evolve primarily due to aberration in neuronal migration, maturation and programming in the development of various topographic zones in the brain, following pathological alterations in glial and neuronal interactions. These are broadly referred as cortical dysplastic conditions. While these dysplastic conditions involving cerebral cortex present as drug resistant seizure disorder, those involving cerebellum present as mass lesions or slowly progressing vertigo.We report 17 cases, representing the histological spectrum of dysplastic, glioneuronal migration disorders which include, hemimegalencephaly (1), tuberous sclerosis (4), Sturge Weber Syndrome with focal dysplasia (1), Dysembryoplastic neuroepithelial tumor (7) and Lhermitte Ductos disease of cerebellum (2). The dysplastic neurons in varied stages of maturation showed neuronal cytoskeletal pathology similar to that in neuro degenerative diseases, especially when associated with cytomegaly. Similarly, cells exhibiting dual expression of glial and neuronal markers were noted in the cerebral dysplastic lesions. The dysplastic glial elements probably form the subependymal giant cell astrocytomas. Dysplastic neuronal elements form the nidus for DNT. When localized, surgical resection ameliorate the symptoms in many of these condition. Study of these conditions provide better insight into glioneuronal interaction and maturation of the brain. Key words : Glioneural migration, Immunohistochemistry. Introduction Abnormalities in neuronal migration or cortical dysplasia (CD) are a heterogenous group of disorders with a variety of subtypes. The term CD has been used in the literature in a broader sense referring to a wide range of abnormalities which grossly vary from complete absence of gyration (agyria or lissencephaly) to polymicrogyria. The spectrum of light microscopic changes associated with CD include i) loss of cortical lamination, ii) marginal glioneuronal heterotopia, iii) single heterotopic white matter neuron, iv) neurons in the molecular layer, v) nodular neuronal white matter heterotopia, vi) balloon cell change, vii) neuronal cytomegaly and viii) cytoskeletal abnormality.1 Cortical dysplasia is frequently associated with various dysgenetic lesions like tuberous sclerosis (TS), neurofibromatosis and Sturge Weber syndrome. A subset of gangliogliomas were also reported to have disorganized cortical architecture.2-4 Hence the presence of co-existent cortical dysplasia in association with these tumors raises important issues regarding their histogenesis. Neocortical development or neurogenesis results from an intricate and interdependent process of proliferation of precursor cells, their migration and differentiation, elimination of supernumery cells by apoptosis, synapse formation and finally cortical remodeling.5 Etiological events of these heterogeneous lesions are also varied and can fit into four main categories namely a) genetic, b) metabolic, c) hypoxic ischemic. and d) toxic environmental factors.6,7 The present study has focussed on contrasting the aberrant lesions with normal cortical development. Material and Methods We retrospectively analyzed 17 patients with history of drug resistant intractable epilepsy of varied duration or vertigo. One case was asymptomatic during life. Nine of them underwent surgery and seven died in status epilepticus with a diagnosis of dysgenetic neuronal disorder. The case of hemimegalencephaly and one case of Lhermitte Duclos disease of cerebellum have been reported earlier.8,9 Surgical and autopsied specimens were fixed in 10% buffered formalin. Representative paraffin embedded sections were routinely stained with hematoxylene and eosin, phosphotungstic acid hematoxylene (PTAH) for glial elements, Bodian and modified Bielchowsky's silver for neural component and luxol fast blue for myelin. Avidin-biotinperoxidase (ABC), or peroxidase antiperoxidase (PAP) immuno-histochemistry was performed on paraffin sections with the following primary antibodies, respective dilution mentioned in the bracket : SMI-31- monoclonal against phosphorylated high molecular weight neurofilament (1:1000), SMI- 33 - monoclonal against non-phosphorylated neurofilament (1:1000), PHF / ubiquitin, monoclonal against paired helical filaments and ubiquitin (1:100), (kindly provided by Inge Grundke-lqbal, Institute for Basic Research in Developmental disabilities, Staten Island, New York; USA), synaptophysin - polyclonal in rabbit (1:50, DAKO), SMI-21, monoclonal against glial fibrillary acidic protein (GFAP, 1:50), Alz-50 - monoclonal antibody to phosphorylated tau (1:10, gift of Dr. Peter Devis, Albert Einstein College of Medicine, Bronx, New York), Neuron Specific Enolase (DAKO, 1:50). As a negative control, the primary antibody was replaced by 3% normal goat serum/ BSA. Section of normal cerebral and cerebellar cortex and frontal cortex from a confirmed case of Alzheimer's disease were incorporated for each antibody, as positive and negative controls. Results A) Relevant Clinical details (Table I) : 17 cases (M:F = 11:6) with age ranging between 6 months to 40 years (mean l8.72 ± 10.88 years) were included in the study. B) Macroscopic features Cortical dysplasia : On sectioning, the cortical gyrus having the dysplastic focus was found to be broad and the cortical ribbon wide, while the adjacent gyri were relatively normal. Underneath the subcortical white matter, an island of grey matter was seen, representing the cortical ectopia (Fig. 1A). Tuberous sclerosis : Cortical tubers appeared as multiple, firm, ill defined broadened cortical gyri, at places as oval areas with central depression leading to broadening of cortical ribbon (Fig. 3). Hemimegaencephaly : The entire left half of the brain was enlarged with marked broading of the gyri and pachygyria. The involved cortex was markedly thickened with obliteration of grey - white distinction. The deep nuclear masses, internal capsule and hippocampal formation were totally disorganized. However the gyral pattern and internal structures on the right side were well preserved. DNT : The involved cortex in most instances were broad and flattened (Fig. 5A) . Gross appearance on cut section was variable and ranged from vague nodularity with greyish avascular jelly like mass to partly cystic ill defined lesion. Lhermitte Duclos Disease : There was diffuse hypertrophy of one half of the cerebellum with gross folial enlargement in both the cases (Fig. 1B). The underlying white matter was poorly discernible. C) Cytoarchitecture and Immunohistochemical profile (Table II) of the lesions Cortical Dysplasia: Both the patients with cortical dysplasia had disarray of cortical lamination. Neurons of varying sizes were present in all the cortical layers. Single neuron or neuronal clusters were present in deep white matter and patchy myelin pallor was noted. Some of these neurons were enlarged and had irregular dendritic processes. These hypertrophied neurons were strongly immunolabelled by antibody to phosphorylated neurofilament protein (SMI -31) (Fig. 2, Inset). An occasional large round ballooned cell with glassy eosinophilic cytoplasm and basophilic cytoplasmic inclusion was also seen (Fig. 2, Inset, lower left). Hemimegalencephaly and Tuberous Sclerosis : The pathological changes in both these conditions were essentially similar, varying in degree and extent of distribution. Cellularity was markedly increased due to the proliferation of neuronal and glial cells. Cortical lamination was obscured due to the presence of haphazardly arranged abnormally large and small neurons mixed with glia. The molecular layer had densely populated large reactive astrocytes as well as bizarre neurons cytologically resembling each other. Grey - white distinction was obliterated.At places, heterotopic neurons were arranged in radial fashion in the subcortical white matter reminiscent of neuronal migration along the radial glia (Fig. 4A). Many of these hypertrophied neurons on Bielchowsky or Bodian silver stained sections showed accumulation of neurofibrillary material in concentric whorls similar to neurofibrillary tangles found in neurodegenerative diseases (Fig. 6A). In the deep white matter multifocal nodular neuronal heterotopia and calcified astroglial scars were seen. Similar to focal cortical dysplasia, these hypertrophied neurons were also strongly immunoreactive to antibodies against phosphorylated neurofilament protein (SMI- 31) indicating their aberrant phosphorylation and dystrophic nature (Fig. 6B). However, they were not immunolabelled with antibodies against phosphorylated tau and ubiquitin. In the white matter occasional large axonal spheroids and dilated dystrophic axons were seen, strongly immunolabelled by antibody to phosphorylated NF (SMI-31) (Fig. 6C). Many of the large neurons also expressed synaptophysin in a granular pattern along the periphery of the perikaryon (Fig. 7A). Apart from cytoplasmic positivity, an occasional neuron had ubiquitin positive elongated intracytoplasmic inclusions (Fig. 7B). Neuronal cells were also immunolabelled with SMI-33, however, the density was never intense. Immunophenotypic nature of the ballooned cells within the cortex and the white matter was ambiguous; some of the cells expressing both neuronal and glial markers on serial sections. Histology of cortical tubers of tuberous sclerosis (TS) was essentially identical to that noted in hemimegaencephaly; only differing in the extent of cortical involvement. They could easily be separated out from the intervening normal cortex. Cortical lamination was typically deranged within the tuber due to extensive gliosis. No neuropil plaques similar to those in Alzheimer's disease were seen. The subependymal giant cell astrocytomas (SEGA), observed in tuberous sclerosis also revealed cytomegaly, similar to neurons, though clustered around ventricles. Similar cytomegaly was not evident in the reactive deep parenchyma, though hamartomatous masses with spotty calcification were noted. Some of the cells of SEGA also showed dual antigenic expression similar to neurons. In the case of Sturge Weber Syndrome, occasional argyrophilic dystrophic neurons were noted at the site of vascular calcinosis in the parieto occipital cortex in addition to heterotropic neurons (2-3 per high power field) and dystrophic neurites in subcortical white matter. Cortical Dysplasia with DNT : Coexistence of cortical dysplasia (CD) and dysembryoplastic neuroepithelial tumor (DNT) was observed in 4 out of 7 cases. On microscopic examination, the lesion was a complex mixture of glial and neuronal cells. Small 'oligodendroglia like cells' (OLC) were the predominant cell population. In most areas they were arranged in columnar pattern with mucinous material in between. Neurons were seen floating within the pools of mucin (Fig. 4B). Corresponding to nodularity seen at low magnification, the OLC were also arranged as nodular aggregates in mucinous background, both in cortical ribbon and grey-white junction. There was minimal nuclear pleomorphism. All the tumors had microcystic background. At places these areas were bordered by capillaries with glomeruloid appearance. Adjacent cortex showed varying degrees of dysplasia. Heterotopic neurons were present in the molecular layer as well as in the subcortical white matter. At places clusters of disorganized neurons were seen in the molecular area of cortical ribbon indicating abnormalities in the tangential migration of the neurons. However, these heterotopic neurons were not labeled by SMI-31 unlike in HME and TS. Synaptophysin immunoreactivity was of variable intensity in the dysplastic areas whereas the adjacent normal cortex showed normal neuropil immunolabelling. The floating neurons were immunoreactive with neuron specific enolase (NSE). Small OLCs within the tumor did not show GFAP, NSE or Synaptophysin positivity. D) Dysplastic lesions of the cerebellum Lhennitte Duclos Disease : Histology of both the cases was more or less similar. The Purkinje cell layer was absent. The granular cell zone composed of large, abnormal neurons had merged with molecular layer. In the molecular layer, unlike in normal cerebellum thick myelinated fibers running horizontally were seen, at places they were vertical. There was irregular gliosis in both the layers. Bodian silver stain confirmed the haphazardly arranged thickened axons in between the abnormal ganglion cells. Immunoreaction using monoclonal antibody for neuronal cytoskeletal proteins showed strong reactivity of the hypertrophied axons and the perikarya of the abnormally large granule cells indicating their dysplastic nature. However staining pattern was variable, like diffuse somatic labeling in some and focal in other areas. In addition, one case showed increased number of calcified vessels in the white matter. None of the abnormal neurons were labeled by tau or ubiquitin antibody. Discussion Cerebral cortical dysplasia is a heterogeneous disorder of cortical development and organization. Inutero migrational abnormalities account for most cases of cortical dysplasia. The histopathological appearance is often varied, making recognition and classification difficult. The present study reviews the histological spectrum of abnormal neurogenesis and their cytoskeletal pathology which would probably explain the temporal evolution of lesions during organogenesis. Of late the term cortical dysplasia had been used to refer microscopic cortical cytoarchitectural abnormalities related to neurons though glial elements also participate in the dysplastic process. As in all these conditions the white matter is also involved, microdysgenesis would probably be the better terminology to designate these conditions.10 The wide spectrum of lesions reflect the time of insults at different points during development. Arrest of migration at different sites depends on the time and extent of damage to radial glia. Prevalence of these conditions in the pediatric age group in association with various dysgenetic lesions in the nervous system suggest abnormalities either in the precursor cell proliferation, migration, glioneuronal - matrix interaction or programming during development. Though various factors have been implicated in the etiopathogenesis, genetic factors still play an important role in early migrational defect which result in severe forms of cortical dysplasia like hemimegalencephaly and tuberous sclerosis. Focal cortical dysplasia or neuronal migration disorder could be due to prenatal vascular etiology.11 Association of CD with DNT has been well documented in literature. These tumors are unique because of their stereotypic clinical, radiological and histological features. Their frequent occurrence in children with preferential location in supratentorial region and little or no cytological atypia suggest their origin during development of neocortex. In four of our seven patients with DNT, focal cortical dysplasia was noted suggesting the origin of these tumors from a dysplastic focus in the cortex. Disorganized neurons in the adjacent cortex with heterotopic neurons in the white matter probably reflect defect in neuroblast migration which continues to migrate postnatally. Clustering of neurons in the cortical ribbon close to surface possibly suggest abnormalities in tangential migration of neurons. Participation of both neurons and glial cells in these tumors without any malignant potential suggest some form of glioneuronal maturational dissociation. There was no cytoskeletal abnormalities in these arrested neurons. However synaptophysin immunoreactivity was variable and light in the dysplastic cortex as opposed to strong neuropil positivity in the normal cortex. This could possibly due to the conformational change in the synaptic vesicle membrane protein in the dysplastic cortex. Possibly DNT represent a tumoral form of cortical dysplasia or neoplastic transformation of a dysplastic focus. Rojiani et al12 found increased number of heterotopic neurons in the temporal lobe white matter compared to frontal and occipital lobes in apparently normal individuals. This could explain the increased frequency of DNT within the temporal lobe. Dumos Duport et al13 suggested the origin of DNT from subpial granular layer which is secondary germinal layer of the developing nervous system, remnants of which can be found in the temporal cortex of the preterm infants. Pathomorphological similarities between cortical tubers of tuberous sclerosis (TS) and hemimegalencephaly (HME) suggest an apparent relationship between the two, the difference being in the extent of cortical involvement. TS is multifocal, frequently bilateral; where as HME is unilateral and diffuse within the affected hemisphere. A defective gene has been identified in patients with TS.14,15 HME may also results from somatic mutation but focal cortical dysplasia being a localized lesion in the midst of normal cortex, is difficult to be accounted for on genetic basis. The most commonly observed pattern of dysplasia in these conditions was disturbed laminar patten of the cortex. The abnormal cortical lamination in cortical tubers of TS and HME most likely reflect a profound defect in neuronal migration in the developing cortex. Defective neuronal positioning could either be due to aberration in neuron - glial attachment or misdirected radial glial fibers. There was significant increase in the number of neurons in the molecular layer in HME and TS patients, probably due to aberrant and persistent migration along the subpial granular cell layer, which normally disappear during development. Hypercellularity was noted in the entire involved cortex in HME. Whether the increased number of neurons resulted from uncontrolled proliferation of precursor cells or reduced programmed cell death (PCD) during development is yet to be answered. During normal development there is 25-50% over production of neuroblasts of which some are destined to undergo physiological programmed cell death (PCD). Though neurons die at all stages during development, PCD affects relatively mature neurons and is governed by trophic signals.16 Another frequent observation was the presence of heterotopic neurons in the white matter either singly or in clusters. Presence of 2-3 neurons per high power field within deep white matter was considered as heterotopic neurons. Presence of single heterotopic white matter neuron could be due to damage of single radial glia, but rowing of neurons in the white matter as seen in HME, suggest tardy migration of a cohort of neuroblasts along the radial glia. Whether genetic factors cause disorganized formation of radial glial fibers or produce defect in the molecules such as laminin that adhere the migrating neuroblast to the glial fibers or defects in the neuroblasts themselves is uncertain. Rorke5 suggested the possibility of defects in genes controlling neuroglial interaction, neuroblast proliferation and PCD, giving rise to neuronal heteropia. Enviommental factors like toxins, germinal matrix hemorrhage and scarring of the ventricular lining by intrauterine infection may also play significant role in the development of neuronal heterotopia.17-19 Neuronal cytomegaly was present in all cases of severe form of CD and appear to be the primary phenotypic derangement. This could be the effect of abnormal neural growth factor or growth factor receptors as suggested by Rorke et al.5 By microfluorometric cytochemical analysis, Manz et al20 showed increase in neuronal DNA (16%) and RNA (40%) content in HME brain compared to normal controls suggesting heteroploidy of chromosomal DNA, enhanced transcription and translation. Kerfoot et al21 have found increased expression of 'tuberin' a tumor suppressor gene protein in the cytomegalic neuron of TS. It is possible that tuberin is also involved with neuronal migration and differentiation. Many of these hypertrophied neurons had intracytoplasmic argyrophilic material and shared a number of antigenic properties with those found in Alzheimer's disease as previously reported by Vinter et al.22 Phosphorylated neurofilament proton and ubiquitin, a component of neuronal inclusions in neurodegenerative diseases were observed in some of the hypertrophied neurons in this study. However they differed from the neurofibrillary tangles of AD in that they were not immunoreactive with antibody raised against paired helical filaments (PHF) and tau. Similar dystrophic neurons containing neurofilamentous inclusion have also been reported by Duong et al.23 Presence of these hypertrophied neurons in different layers of the cortex probably suggests dystrophic change due to biochemical alterations in abnormal location. Interestingly one of our patients with TS had long standing dementia, which has not been reported in literature. Hence the significance of these cytoskeletal pathologies though uncertain, still could represent an intermediary stage of neuronal degeneration, affecting the cognitive function variably. Synaptic dysgenesis also plays an important role in the development of cortical dysplasia. By morphomertic study O'Kushy et al24 showed significantly increased number of synapses in a radial column of cortex though density of synapses per unit volume of cortex did not differ significantly from the controls. Abnormalities in synaptic circuits due to malpositioning of the neurons or biochemical alteration within the neuronal circuitry could be the cause for seizure in these patients. Many of the ballooned cells of the TS and HME showed variable positivity with GFAP and neurofilament protein. This dual positivity suggest a failure of the cells to commit to a specific' phenotype, implying pleuripotent or dedifferentiated nature of these cells. Ballooned cells associated with SEGA also showed similar immunohistochemical pattern, while others found a much higher proportion of GFAP positive cells in cortical tubers than SEGA. This would probably suggest that acquisition of GFAP is needed for migration and maturation.25,26 Histological and immunohistochemical findings of Lhermitte Duclos Disease suggest the dysplastic nature of this lesion.27 But the reason of dysplastic change in the granule cells and origin of hypermyelinated fibers in the superficial layer of the cerebellar cortex and projection areas are not clear. It is possible that there is a defect in cytoskeletal protein metabolism with accumulation of phosphorylated neurofilament protein in the perikarya leading to cytomegaly. Thus a wide spectrum of cortical dysplasia reflects derangement of the normal process of neocortical development. Cortical dysplasia may be the result of multiple events and manifest in several phenotypic ways, brunt of the insult being on neuronal migration. Role of subplate neurons in cortical remodeling, different morphoregulatory molecules, like cell adhesion molecules and surface adhesion molecules modulating the neuron-neuron, neuron - glia or extra cellular matrix interaction and soluble trophic factors, probably play a role singly or in concert in the evolution of these lesions. Hence high prevalence of these lesions in children, the early onset of seizures and varied morphological patterns probably represent some form of dissociation in glio - neuronal interaction and neuronal migration during development. Identification of these lesions especially DNT, focal cortical dysplasia and LDD has theraputic and prognostic implications as the deleterious long term effects of aggressive chemo or radiotherapy with a mistaken impression of neoplasia can be avoided in these young patients. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02120f4.jpg] [ni02120f7.jpg] [ni02120f3.jpg] [ni02120t2.jpg] [ni02120f1.jpg] [ni02120f6.jpg] [ni02120t1.jpg] [ni02120f2.jpg] [ni02120f5.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}