 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
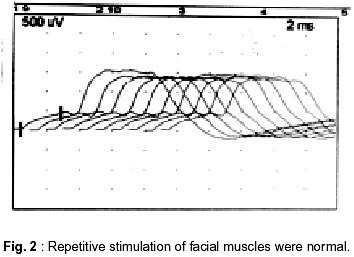
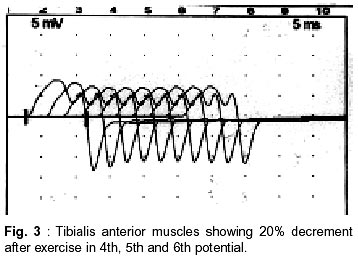
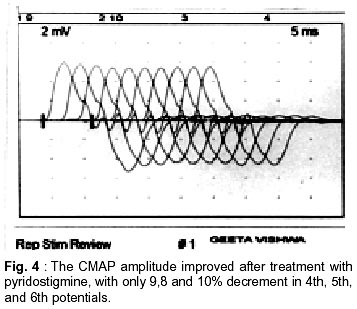
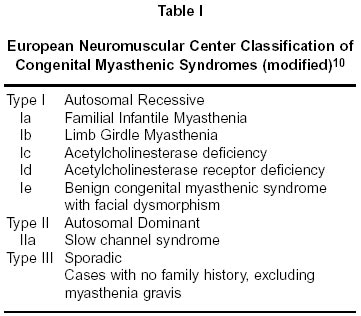
Neurology India, Vol. 50, No. 4, Dec, 2002, pp. 500-503 Case Report Autosomal Recessive Limb Girdle Myasthenia in Two Sisters A. Shankar, T. Solomon, T.P. Joseph, C. Gnanamuthu Department of Neurological Sciences,
Christian Medical College and Hospital,
Vellore - 632 004, India. Accepted for publication : 21st January, 2001. Code Number: ni02134 Summary Limb girdle myasthenic syndromes are rare genetic disorders described under the broad heterogeneous group known as congenital myasthenic syndromes and present with mixed features of myasthenia and myopathy. The familial limb girdle myasthenia has been described as one with selective weakness of pectoral and pelvic girdles, showing a positive response to edrophonium chloride. A report of two sisters affected by this disorder is presented. Key words : Familial, Limb-Girdle, Myasthenia. Introduction Congenital myasthenic syndromes (CMS), of which limb girdle myasthenia is a variant, are a heterogeneous group of autosomal recessive disorders affecting neuromuscular transmission. The presentation may vary from mild limb weakness to severe disability with life threatening episodes. These syndromes usually spare the cranial musculature. In recent years these syndromes have been investigated in detail and have been classified into a number of subgroups that can be recognized by the clinician.1,2 The diagnosis can be confirmed with reasonable certainty with the help of meticulous clinical examination and electrophysiological studies. Even in centers where advanced investigations are unavailable, basic electrophysiology with negative acetylcholine receptor antibody will help to identify this condition, especially in the presence of a classical clinical presentation. It is mandatory to rule out polymyositis, stiffman syndrome, myopathic disorders and neuromyotonic disorders.3 We report here two sisters who presented with progressive weakness of the limb girdles, with pelvic girdle being more affected than the pectoral. These cases are presented for their rarity and the difficulty encountered in diagnosis, as they are easily confused with familial myopathies. Case Report A 22 year old lady from Bhutan, born of non- consanguineous parents, presented with severe weakness of proximal limb muscles that was induced by moderate exercise. The illness had begun at the age of 4 years. The weakness was predominantly in the pelvic girdle, and she developed buckling of knees and waddling at the hips after walking for ten minutes. She also had weakness in the lower limbs after climbing 2-3 flights of stairs or walking a kilometer. There was fatiguability on prolonged chewing. Despite the long-standing presence of all these problems, she had never developed any respiratory distress, diplopia, ptosis or nasal regurgitation. Clinical examination was normal at rest. No facial dysmorphism was noted (Fig 1). Gait was normal after routine activity. After climbing three flights of stairs, she developed a waddling gait and bilateral foot drop. The muscle tone was normal and there was no wasting or fasciculations. There were no sensory deficits. Deep tendon reflexes were normal and plantars were bilaterally downgoing. There was no demonstrable fatiguability involving the ocular, pharyngeal, facial or tongue muscles. However, after chewing for a long period, she developed mild weakness of the jaw. Routine blood examinations were normal. Liver function tests were within normal limits and serum creatine kinase (CK) was 57 IU/L. Serum lactate done before and after exercise showed a normal rise in lactate levels. Anti-nuclear antibody, anti-double stranded DNA and rheumatoid factor was negative. Nerve conduction studies and needle EMG were normal at rest. Slow repetitive supramaximal stimulation in the deltoid, biceps, facial muscles, tibilias anterior and the abductor pollicis brevis were normal at rest (Fig. 2). However, after the patient climbed a flight of stairs 3 times in quick succession, the recording showed a 20% decrement in the tibialis anterior and 27% decrement in the abductor pollicis brevis (Fig. 3). After treatment with pyridostigmine, the CMAP amplitude improved and the percentage of decrement reduced. Repetitive CMAPs were not noted (Fig. 4). A routine muscle biopsy did not show any myopathy or tubular aggregates in the muscle. Electron microscopy of the muscles did not reveal any abnormality in the myocyte or neuromuscular junction. The acetylcholine receptor antibody was negative. The weakness noted on exercise improved remarkably with intravenous administration of 10 mg of edrophonium chloride. Therefore, she was started on pyridostigmine (30 mg three times a day), following which there was significant improvement over a period of 2 weeks. She was then able to climb a flight of stairs seven times, beyond which she became symptomatic again. Her younger sibling, who was 18 years old, had almost the same clinical picture. Her EMG, which was normal at rest, showed decremental response following strenuous exercise and she too had a similar response to edrophonium chloride. Hence, she too was started on pyridostigmine, with considerable clinical improvement. No other family member had this problem. Discussion Congenital myasthenic syndromes are known to have pre-synaptic, synaptic or post-synaptic defects.1,2,4 CMS usually presents within the first 2 years of life, but they may present even in adulthood. Since there is very little difference between the various phenotypes of CMS, differentiation usually depends on detailed evaluation, including electrophysiological and molecular genetic studies.5-7 Initially these cases were considered as a congenital myopathy with myasthenic features. The first case of a pure limb girdle myasthenia was reported in 1966.7 This report concerned a brother and a sister, both of whom were in the second decade of life and suffered from myopathic weakness, most prominent in the extremities. There was no associated oculo-bulbar or facial involvement and no muscular atrophy. Ocular muscle involvement may be minimal or absent in slow channel syndrome, neuromuscular junction acetylcholine esterase deficiency and familial limb girdle myasthenia.8 These patients had slightly elevated CK levels and EMG features suggestive of myasthenia, including improvement with edrophonium chloride injection. The limb girdle myasthenia has been variously classified by Engel et al9 as a partially characterized syndrome and by the European Neuromuscular Center International Workshop as an autosomal recessive syndrome type 1b (Table I)10. Lecky et al11 described a patient who had weakness without the classical myasthenic fatiguability. The face and proximal limb muscles were more affected than the distal muscles. Muscle biopsy was reported as normal in this case. There was no improvement with edrophonium chloride, but some improvement followed administration of 3,4-diaminopyridine. Vincent et al12 also reported a similar patient who improved following treatment with 4-aminopyridine. Gardner-Medwin13 reported a series of 27 cases from Newcastle, characterised by similar gait disturbances and lack of myasthenic (diurnal) fatiguability. In their series, the gait disturbances fluctuated gradually over weeks or months, for no obvious reason. The post exercise weakness often lasted for many days. Some of their patients improved with small doses of pyridostigmine, while one patient had sudden worsening. Vasant and Sathyanaraynaswamy14 reported six cases of insidious onset of proximal muscle weakness, without ocular or bulbar involvement. Detailed studies since 1990 have identified the DNA sequence encoding for the various subunits of the acetylcholine receptor gene (alpha, beta, delta and epsilon subunits in adults and gamma subunit in the fetus), and researchers have succeeded in cloning the catalytic and collagenic tail (ColQ) subunits of asymmetric acetylcholinesterase.15 The knowledge gained from these experiments has been utilized to identify the defects of neuromuscular junction, which are responsible for the various congenital myasthenic syndromes. Various researchers have identified over 56 mutations of the AchR subunit gene so far. These include slow channel mutations, fast channel mutations, frame shifting rearrangements and null mutations. A vast majority involve the epsilon subunit of AchR.15,16 Both our patients had features of proximal and distal muscle involvement on exertion without oculopharyngeal involvement. Recovery occurred within an hour on resting. The response to intravenously administered edrophonium chloride and to oral pyridostigmine was good, as was the experience of other authors.7,11 It is noteworthy that on repetitive stimulation, no decrement was seen at rest, but a significant decrement was seen after exercise. The diagnosis of limb girdle myasthenia was made on the basis of clinical manifestations and EMG findings. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02134t1.jpg] [ni02134f1.jpg] [ni02134f4.jpg] [ni02134f2.jpg] [ni02134f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}