 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
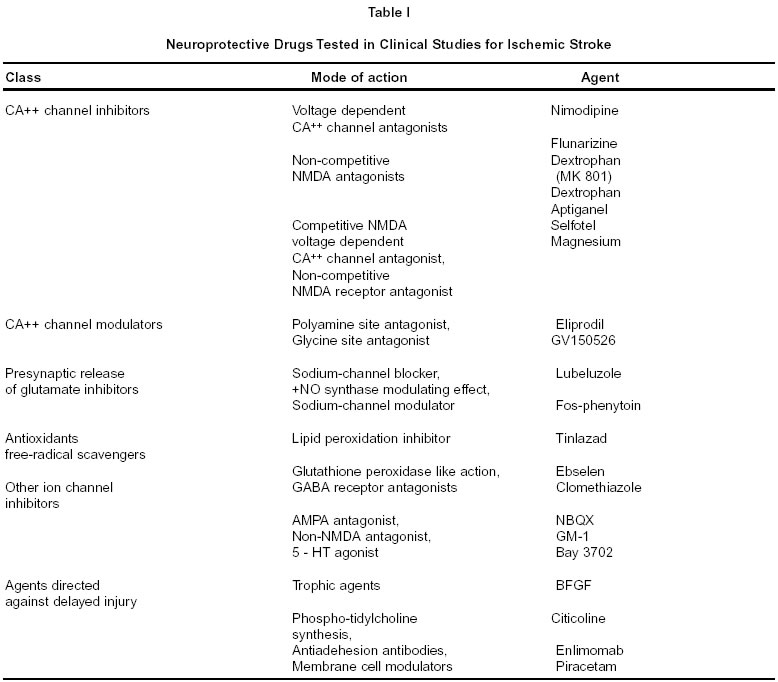
Neurology India, Vol. 50, (Suppl. 1), Dec, 2002, pp. S57-S63 Neuroprotection for Acute Ischemic Stroke : An Overview S. Mohandas, J. Mani, R. Borgohain, S. Sitajayalakshmi Department of Neurology,
Nizam's Institute of Medical Sciences,
Hyderabad - 500 082, India. Code Number: ni02163 Summary The introduction of thrombolytic therapy has not only injected fresh optimism in stroke management, but has also given a fillip to stroke research, and spurred a number of clinical trials in stroke therapy aimed at salvaging potentially viable ischemic brain tissue. Though a large number of neuroprotective drugs are successful in experimental animal models they have not translated to effective clinical therapy due to a variety of reasons. This has led to a lot of introspection on the methodologic issues in stroke trials and also led to better understanding of ischemic brain damage. It may be realistic to expect that the advances in understanding would evolve into effective neuroprotective therapies in the future. Key words : Neuroprotection, Acute ischemic stroke. Introduction The introduction of thrombolytic therapy for acute ischemic stroke has been a historical breakthrough in stroke management in this millennium. It has injected a new sense of optimism hitherto unknown in the history of stroke management. This has motivated hectic activity among stroke researchers and spurred a number of clinical trials in stroke therapy in an attempt to salvage potentially dying brain tissue. The ischemic penumbra Ischemic cell injury is not an instantaneous event. A complex series of physiological, biochemical and molecular cascading mechanisms lead to cell death following ischemia. A thorough understanding of this complex and dynamic mechanism is a prerequisite for any effective neuroprotective strategy in acute stroke. Normal cerebral blood flow is about 50-60 ml/100g/min. When blood flow falls below 10-15 ml/100g/min it triggers off the ischemic injury cascade. Experimental studies of focal brain ischemia modeled in rats, cats and non-human primates, suggest that after an ischemic insult the neurological damage spreads circumferentially from the core of the infarct,1 and it gradually enlarges over 3-4 hours in rodents and 6-8 hours in non-human primates.2 The ischemic penumbra is the area of cerebral tissue surrounding the infarct core in which metabolic activity is suppressed, physiological processes are altered, but destruction is not inevitable. The ischemic penumbra is the area which is the focus of all neuroprotective strategies in ischemic stroke. Therapeutic strategies aim at interruption of the ischemic cascade. Positron Emission Tomography (PET) studies in experimental animals can differentiate ischemic from infarcted tissue, i.e. identify the ischemic penumbra. Ischemic but viable tissue has been demonstrated for up to 48 hours after the acute insult.3 The ischemic cascade Critical reduction in cerebral blood flow deprives brain of substrates (glucose and oxygen), and within minutes the energy demands exceed the capacity of the brain to generate adenosine triphosphate (ATP). There is tissue acidosis due to lactate excess, which triggers off a failure of ionic pumps, resulting in the generation of free radicals, neurotransmitters and inflammatory mediators. The complex cascades of events disrupt cell homeostasis and ultimately cause cell death. Targets for neuroprotective therapy Many neuroprotective drugs have been developed in the last few years, which act on specific targets involved in the presumed ischemic cascade. Glutamate : The ability of glutamate exposure to trigger central neuronal death has been known for over 40 years and substantial evidence for the role of glutamate in mediating ischemic brain injury has accumulated over the past 10-15 years.4 Ischemic neurons deprived of oxygen and glucose rapidly lose ATP and become depolarized, leading to the synaptic release of transmitter glutamate from the depolarized astrocytes. The excess glutamate stimulates the a Amino 3 hydroxy 5 methyl 4isoxazolepropionic acid (AMPA), N Methyl D Aspartate (NMDA) and kainite receptors with consequent influx of calcium and sodium through channels gated by these receptors. The NMDA receptors : The NMDA receptors mediate rapidly triggered neurotoxicity whereas kainite receptors induce neurotoxicity at a slower pace. The crucial role of NMDA mediated neurotoxicity in ischemic brain injury was corroborated by the reduction in the infarct volume in animal models of brain ischemia in many laboratory studies using NMDA antagonists.5 However, despite evidence of reduction in infarct volume in animals most drugs like Selfotel and Cerestat have had to be abandoned due to undesirable side effects or poor clinical benefit in humans.6,7 Magnesium blocks the voltage gated ion channel of the NMDA complex and acts as a non-competitive NMDA antagonist at higher doses. It may inhibit calcium influx into injured neurons. Pilot studies have shown encouraging results.8 Clinical trials with magnesium are in preparation. AMPA and Kainate receptors : Several factors during tissue ischemia like lactic acidosis, free radical accumulation and the inhibitory effects of calmodulin may under-regulate the NMDA receptors and increase the contribution of AMPA and kainite receptors in ischemic injury. Inhibitory Gamma Amino Butyric Acid (GABA) containing cortical neurons and oligodendrocytes are specific cell types that may be selectively vulnerable to AMPA/kainite induced cell death. Drugs that act on AMPA/glycine receptors are in phase II/III trials. Clomethiazole is a GABA agonist, which gates the chloride channel. It showed no overall benefit in a recent phase III trial but improved the functional outcome in patients with large cortical infarcts.9 Calcium : Calcium homeostasis is crucial to cellular integrity. It is maintained by regulation of calcium influx at voltage and ligand gated calcium channels (mediated by glutamate), by ATP dependent calcium exchange pumps and buffering of calcium ions by various proteins like calmodulin. During cellular ischemia there is an increased inflow of calcium ions through the gated channels due to failure of the energy dependent pumps, which leads to an excess of calcium in the cell. Raised intracellular calcium accelerates many potentially injurious processes.10 It activates various enzymes like phospholipases, which hydrolyse membrane bound glycerophospholipids and facilitates perodixation of membrane bound lipids. Calcium also activates cellular proteases that lyse structural proteins and initiates free radical mechanisms. Nimodipine blocks the voltage gated calcium channels. In the early trials, nimodipine had shown significant functional improvement in patients when used within 12 hours of stroke onset. This led to the VENUS (Very Early Use of Nimodipine in Stroke) trial in Netherlands in which the drug was administered within 6 hours of stroke onset. However, a subsequent Cochrane review of the role of calcium antagonists in acute ischemic stroke was unable to reproduce the positive results of the earlier trials. Calcium antagonists were shown to have no effect on the poor outcome in acute ischemic stroke. Intravenous nimodipine and larger doses of the drug were associated with poorer outcome. Subgroup analysis of oral nimodipine used within 12 hours was not beneficial.11 These findings led to the premature termination of the VENUS trial. The VENUS trial also did not demonstrate a beneficial effect when nimodipine was used early in stroke patients.12 Reperfusion injury Thrombolysis or spontaneous recanalisation leads to tissue reoxygenation and formation of free radicals and the influx of inflammatory cells. Free radicals Free radicals and depletion of endogenous antioxidants have been shown to aggravate ischemic injury in animal models. They peroxidate various molecules, disintegrate structural proteins and inactivate various enzymes. This can lead to cell necrosis or trigger apoptotic processes. Antioxidants scavenge free radicals and reduce cellular and vasogenic edema and favour reestablishment of cellular homeostatis. Antioxidants with potential protective effects include glutathione, vitamin E and ion chelators. Tirilazid mesylate is one such drug, which entered phase III trials but showed conflicting results in two different phase III trials.13 Ebselen, when given orally in patients within 48 hours of stroke onset demonstrated benefit at 30 days.14 Citicoline is an antioxidant, which may act through delayed restorative mechanisms. The drug was given within 24 hours and showed significant improvement in early studies but a recent large trial showed it to be safe but ineffective.15 Nitric oxide Nitric oxide (NOS) is a free radical substance with several established physiological properties when produced in small concentrations. These include regulation of vascular smooth muscle cells, neurons, macrophages, platelets and neutrophils. Nitric oxide exists in two isoforms, the endothelial and the neuronal isoforms. Increased endothelial isoform activity appears to be protective in cerebral ischemia probably because of its cerebral vasodilatory effects and its inhibitory effects on platelet aggregation and leukocyte adhesion. Mouse models that lack endothelial NOS develop significantly larger infarcts after permanent arterial occlusions.16 In contrast to the neuroprotective effects of enhanced endothelial NOS, neuronal NOS released during cerebral ischemia is a powerful oxidant and highly neurotoxic. It peroxidates lipids and disrupts mitochondrial enzymes and nucleic acids. It can trigger apoptotic cell death.17 Inducible or immunological NOS, another form of NOS appears in a delayed manner after a time lag of 6-12 hours after the onset of cerebral ischemic injury. It is a cytokine, which is shown to contribute to delayed ischemic injury. Unlike the earlier two isoforms it is only a product of damage and does not have physiological functions. Targeting this isoform may therefore have the advantage of a neuroprotective effect without dangerous side effects. There may be potential for damage in targeting the earlier two isoforms by virtue of disruption of their important physiological effects. Aminoguanidine is one such potential agent, which inhibits immunological NOS. It is known to be safe for human use and also provides a wider time frame of potential efficacy as it targets the delayed isoform of NOS. It is now ready for clinical trials. Inflammatory mediators There is growing evidence that inflammatory mediators and the immune system are activated after cerebral ischemia. Cytokines and chemokines released in ischemic tissue attract inflammatory cells to the injured tissue. Leukocytes can cause secondary damage by free radical formation, release of cytotoxic enzymes, alteration of vasomotor reactivity, vascular permeability and microvessel occlusion. Microvessel occlusion may persist even if the larger vessel has been recanalised by thrombolysis. Enlimomab Neutrophils have a role in the development of cerebral infarction and mediate some aspects of reperfusion injury. When blood flow is restored on reperfusion, the leucocytes bind to intercellular adhesion molecules (ICAM) on microvessel walls leading to microvessel occlusion that is not thrombolysis sensitive. Enlimomab is a monoclonal antibody directed against ICAM. It was given intravenously within 6 hours (160 mg) of stroke followed by 40mg/day from day 2-5 in 625 patients. But the results were negative with worse outcome and increased mortality in the treatment group because of infections and fever.18 Zinc There is increasing suspicion of a pathogenic role of zinc in selective neuronal loss after transient global ischemia. Zinc is an important component of various metallo-enzymes and transcription factors and also serves as a neurotransmitter / neuromodulator in the central nervous system. The exact role of synaptically released zinc is not clear. The concept that zinc neurotoxicity might contribute specifically to ischemic brain damage was first raised by Tonder.19 The possibility that synaptically released zinc may be analogous to glutamate has been raised. There are other indications that the first step in zinc mediated neuronal death, like calcium mediated neuronal death is the excess entry across the plasma membrane - probably facilitated by various voltage and receptor gated channels. Zinc can induce apoptosis or necrosis depending on the intensity of exposure. Ischemic apoptosis Apoptosis refers to genetically programmed cell death wherein cells die with minimal inflammation or release of genetic material. In its pure form such as during development or after ionizing radiation, apoptosis is characterized morphologically by coarse regular chromatin condensation, loss of cell volume and extrusion of membrane bound cytoplasmic fragments (apoptotic bodies). These features of apoptosis can be found in neurons and glia after ischemic injury, but is usually admixed with additional features of cell necrosis. Growing evidence now indicates that programmed cell death may also play a significant role in neuronal death after focal or global ischemia.20 Programmed cell death is mediated by a group of cysteine proteases called capsases. Activation of capsase 3, which bears homology to the death promoting enzyme CED 3 has been demonstrated in the cortical and striatal neurons several hours after focal ischemia.21 The best evidence implicating apoptosis, as a mechanism in ischemic brain cell death is the protective action of genetic or pharmacological interventions aimed at selectively blocking the apoptosis cascade. Transgenic over-expression of certain anti apoptotic genes like bcl-2 was found to reduce infarct volume in mice subjected to focal ischemia. Necrosis or apoptosis? It is speculated that apoptosis and necrosis may be triggered parallel in the brain following ischemia. The severity of the ischemia and the consequent excitotoxic injury, and neuronal maturity may determine which process predominates. Low levels of excitotoxic exposure causes cultured neurons to undergo apoptosis but when the same system is exposed to more intense excitotoxicity, it undergoes necrosis. Intracellular calcium concentrations appear to be critical for neuronal survival. Calcium overload causes cell necrosis, whereas paradoxically calcium starvation results in neuronal apoptosis. Calcium overload and starvation area extreme states may be triggered by the same ischemic event separated in space and time. Calcium overload may prevail in the acute stage, close to the ischemic core and cause cell necrosis, but further away from the core and at later time intervals, calcium starvation and apoptosis may predominate in certain cells.22 Other neuroprotectors with potential clinical interest Lubeluzole is a sodium channel blocker that prevents presynaptic release of glutamat. Lubeluzole showed promise in animal models and showed positive results in a phase II trial in Europe. But two-phase III trials done in Europe and in United States23 showed conflicting results, with the European24 group reporting completely negative outcome. Fosphenytoin is a widely used anticonvulsant agent and is currently in phase III trials. It is a sodium channel modulator that reduces the presynaptic release of glutamate. Piracetam present in phospholipid membranes helps maintenance of membrane bound cell functions including ATP production, neurotransmission and secondary messenger activity. Phase III trials have shown marginal benefits in a small subgroup of patients25, but a recent meta analysis showed slightly adverse effect of piracetam on early death with no benefit in functional outcome in patients with acute stroke.26 Hypothermia has long been recognized as a potential approach to cerebral protection during cardiac procedures. Apart from reducing metabolic demand, hypothermia reduces excitotoxicity and may have other cellular protective effects. Conversely hyperthermia in stroke patients may exacerbate neural injury by several mechanisms viz. neurotransmitter release, free radical production, blood brain barrier changes, ischemic depolarization and cellular degradation. Moderate hypothermia (30-35oC) has shown to reduce infarct size in temporary focal ischemia but severe hypothermia has caused detrimental effects in primate stroke models. Calpain (calcium dependent cysteine protease) inhibitors, opioid antagonists and serotonin antagonists have antiischemic effects and warrant evaluation. The reader is referred to table 1 for summary of drug trials. Current status of clinical trials in neuroprotective therapies The search for drugs to protect cerebral tissue has consumed enormous resources over the past few years. A broad spectrum of compounds with disparate mechanisms of action has been considered. Quite a few have shown significantly favourable results in animal models, but not a single one of them has stood the test of clinical trials in patients of stroke. Several drugs that initially appeared promising have been subsequently abandoned either because of lack of efficacy or unsatisfactory risk benefit ratio. Why have neuroprotective agents failed by the bedside? Many drugs that have been selected after rigorous laboratory experiments showed significant reduction in infarct volume in experimental animals. But, not a single drug has been able to withstand clinical trials in patients and show reasonable benefit with tolerable side effects. Why the positive laboratory results were not translated into success at bedside has several explanations. Are animal models useful? There is no doubt that laboratory and clinical models of investigation have unique and complementary roles in neuroprotective drug research. Preclinical studies on animal models are an indispensable tool to probe into the molecular and cellular basis of ischemic neuronal death. They have helped in understanding the key concepts of ischemic injury via ischemic penumbra and ischemic apoptosis. The reproducibility of the results and quantification of injury in the form of infarct volume in animal models provide experimental advantage. However, it is the same constancy and uniformity of the experimental setting that makes translation to the complex and variable clinical situation imperfect and difficult. Difficulties in translation of animal studies to patients of stroke:
What are the solutions? Identifying specific markers to define potentially salvageable tissue Diffusion and Perfusion weighted magnetic resonance imaging can identify tissue at risk and compare the pre and post treatment changes in these variables rather than use delayed outcomes like functional recovery at three months which may be influenced by many other factors. A surrogate marker may be used at the time of drug delivery to confirm that the desired effect is actually obtained. For example, functional imaging techniques like Magnetic Resonance Spectroscopy may help confirm that a particular drug is producing the desired biochemical effect. Use more robust methods of evaluation of outcome for e.g. a combination of various stroke outcome scales or a binary outcome of a continuous scale. Stratifying stroke patients according to stroke type and severity Severity of stroke has a direct relation to the residual disability in patients with stroke. Strokes of varying severity may also have a nonlinear profile of recovery and those at the lower end may show a greater recovery in the early phase. However, in most trials patients with any stroke severity are enrolled into nonstratified groups and randomized to receive either medication or placebo and expected to recover uniformly. Hence severity of the baseline disability should be taken into account during the stratification and randomization for drug trials. Using combination therapies Use of combination therapy of a thrombolytic agent with a neuroprotective drug may ensure better delivery of the latter to the ischemic potentially salvageable tissue. The administration of a combination of neuroprotective drugs in the form of a neuroprotective cocktail may also be considered. Combinational approach may yield greater neuroprotection than each approach alone and may permit a longer therapeutic window. Neuroprotective synergy has been demonstrated in experimental animals where the beneficial effect of the combination was better than either drug alone (dextrophan - NMDA antagonist with cycloheximide an antiapoptotic drug). Use of combination NMDA and AMPA antagonists can increase overall antiexcitoxicity on ischemic neurons. The problematic side effects of pan blockade of receptors like respiratory depression can be overcome by the use of subtype specific drugs. An alternative method may be to reduce glutamate release, hypothermia, GABA agonists or sodium channel blockers. Trying novel approaches These include targeting glial tissue and use of other novel methods like calpain inhibition and antisense oligonucleotides that modify gene expression. There is more recent evidence that the ischemic penumbra in humans remains metabolically viable for up to 24 hours after the onset of cerebral ischemia. It may be possible to target the delayed neurodestructive pathways like free radical induced oxidative damage, cell energy depletion and altered gene expression that lead to apoptotic cell death. Conclusion Although no neuroprotective agent is yet available for acute stroke therapy, the information gained from the clinical trials is invaluable. We know that the agents are biologically safe. We also understand the limitations of preclinical models. This information will hopefully help researchers design more effective trials for the future. References
Copyright 2002 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni02163t1.jpg] |
| |||||||||

{kind=link}