 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 51, No. 1, Jan-Mar, 2003, pp. 104-109 Review Article Current status of osmotherapy in intracerebral hemorrhage J. Kalita, P. Ranjan, U. K. Misra Department of Neurology, Sanjay Gandhi PGIMS,
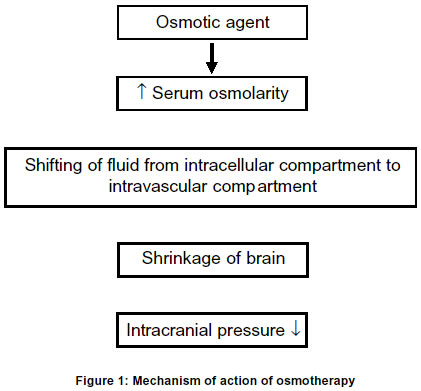
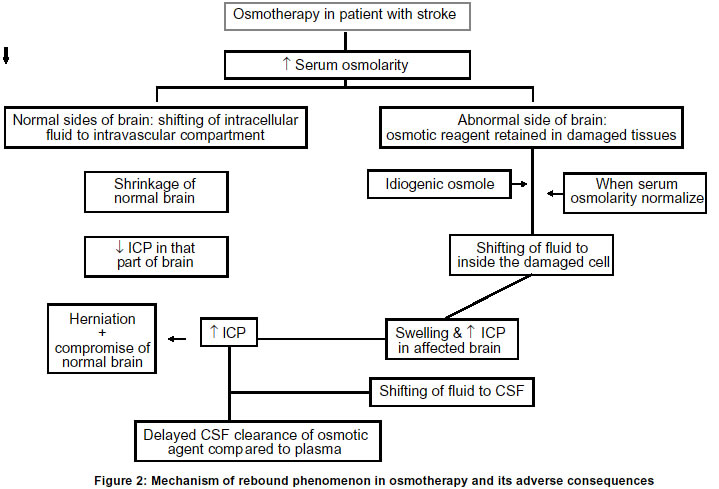
Lucknow, India. Code Number: ni03031 Intracerebral hemorrhage (ICH) is defined as bleeding into the brain parenchyma that may extend into the ventricles and in rare cases into the subarachnoid space. It is the most common form of hemorrhagic stroke representing 10-15% of all strokes and results in more deaths or major disability than due to cerebral infarction. Only 38% of affected patients survive one year.1 The annual incidence of ICH is approximately 10-20/lac individuals and has remained essentially unchanged over the past two decades. The reported incidence of ICH among the Asian population is higher than among the whites in USA and Europe. Advancing age, hypertension, alcoholism, low serum cholesterol, amyloid angiopathy, arteriovenous malformation, ruptured aneurysm, coagulation disorder and drug abuse are important risk factors for spontaneous ICH.2 Earlier, hypertensive ICH was considered to be a monophasic event, but lately it has been shown by CT scan that hematoma can slowly expand over time and can even be recurrent.3 In a study of 103 patients with ICH, the hematoma expanded in 26% patients within 1 hour and in another 12% within 20 hours.4 The enlargement of hematoma has been attributed to continued bleeding, mechanical disruption of surrounding vessel, acute hypertension or a local coagulation deficit. Mechanism of raised intracranial pressure The presence of hematoma initiates edema and neuronal damage in the surrounding parenchyma. Fluid begins to collect immediately around the hematoma resulting in edema which usually persists for five days,5 but may last for two weeks. Early edema around the hematoma results from the release and accumulation of osmotically active serum protein from the clot.6,7 Cytotoxic and vasogenic edema follow, owing to failure of the sodium pump, death of neurons and disruption of the blood brain barrier.8 Mass effect as a result of the volume of the hematoma, edema surrounding the hematoma and obstructive hydrocephalus may subsequently result in midline shift and herniation. This is the major cause of death during the first few days after ICH. The local increase in volume following ICH is initially accomodated by ventricular and subarachnoid spaces and later a marked progressive elevation of intracranial pressure (ICP) is seen, especially in patients with massive ICH. Localized mechanical damage and even transtentorial herniation may be seen in the absence of global increase in intracranial pressure.9,10 The major cause of mortality in ICH in acute stage is raised ICP. Various medical and surgical procedures have been undertaken to tide over this problem. In this review, the current status of osmotic therapy will be discussed. Mechanism of osmotic agents in ICP reduction The physiological principle underlying the ICP-lowering effect of various osmotic agents is the same. The solute must be relatively restricted in its entry across the blood brain barrier. An osmotic gradient is necessary to draw water from the brain cell to the site of higher osmolality, the plasma (Figure 1).11,12 Once the solute has reached equilibrium in both plasma and brain cell, the intracellular volume returns to its initial state and the intracranial pressure returns, to its initial level. Dimattio et al in 1975 showed in cats that a minimum of 10 per cent change in serum osmolality, i.e. about 30 mosm/L was needed to increase or decrease the water content of the brain.13 However, smaller osmotic gradients have been shown to provide effective therapy in man. In a clinical study of Glycerol therapy, Rottenberg et al in 1977 reported that an osmotic gradient of just 10 mosm/L, was effective in reducing the intracranial pressure.14 In addition to its complex effects on brain volume and metabolism, hyperosmolality in laboratory animals has also been shown to inhibit the formation of CSF within the ventricular system.11,13 The data on human beings regarding the quantitative importance of such an effect, however, is lacking. Idiogenic osmoles: Osmotic gradients between brain and plasma are sustained only for a brief period. Osmotically active solutes appear in the brain to increase the intracellular osmolality as an adaptive response to equal the increased osmolality of serum. These unidentified intracellular osmoles have been termed as "idiogenic osmoles" i.e. osmoles of undetermined etiology.15 These are transient in nature i.e. these appear rapidly in response to a steep osmotic gradient, but subsequently these are no longer detected as the intracellular and extracellular osmolality equalizes. While the appeareance of some of the unidentified osmoles is attributed to an increase in the intracellular concentration of free amino acids, it accounts only for a small part of the total idiogenic osmoles.16 An increase in the ionic dissociation of intracellular potassium from a bound to free state may also account for the transient appearance of idiogenic osmoles. The efficacy of osmotherapy in reducing the ICP depends upon the presence of an osmotic gradient between blood, CSF and brain. Once the osmotic gradient has been obliterated by the entry of the solute into the CSF and brain compartments, and by the transient appearance of idiogenic osmoles, the therapeutic efficacy of osmotherapy is lost and rebound rise in ICP may develop. Mechanism of rebound: Rebound phenomenon of osmotic agents is the most limiting factor of their use. The following mechanisms are thought to be responsible for rebound phenomenon (Figure 2).
Osmotic agents used in osmotherapy The introduction of osmotic agents in the treatment of raised ICP was initiated by experiments of Weed and McKibben in 1919 which showed the ability of intravenous hypertonic solutions to acutely lower ICP.17 Hypertonic glucose (50 ml of 50 gm/dl) intravenously was first used in clinical practice. However, it was soon recognized that after an initial drop in ICP, a secondary rise i.e. a rebound in intracranial pressure occurred as the solute rapidly reached equilibrium in CSF and brain. Hypertonic sucrose had a brief vogue in clinical practice during the 1930s, but the occurrence of crystalluria and hematuria limited its use.18 Intravenous albumin was found to have no clinical usefulness in reducing the ICP. During 1960s and 1970s hypertonic urea and glycerol were widely used, but in recent years mannitol has become popular. Various osmotic agents used to lower raised ICP are discussed below. Urea:The ability of hypertonic urea to reduce the intracranial pressure was first studied by Fremont-Smith and Forbes in 1927; although it was introduced in clinical practice only in 1956. Urea is a potent dehydrating agent because of its low molecular weight (60 Dalton), slow elimination from the blood and relatively slow penetration of the blood brain and blood-CSF barrier.19 Sterile lyophilized urea in 10% invert sugar, or 5 to 10% dextrose was given intravenously as a 30% weight-volume solution. Dosage of 1.5 gm/kg of body weight was recommended but in the elderly, 0.5 gm/kg is preserved. Following urea therapy, a rebound rise in ICP occurs, particularly when it is administered in excess of the urinary excretion. Urea, moreover, is not inert; it is epileptogenic hence its use is not recommended. Glycerol:Glycerol is a trivalent alcohol and has been used both systemically and orally to reduce raised ICP.20 Unlike mannitol, glycerol is not metabolically inert. It is partially metabolized to carbon dioxide and water. Glycerol given orally or by nasogastric tube, may reduce ICP within 30 to 60 minutes. 1.2 gm/kg is administered initially followed by a maintenance dose of 0.5-1 gm/kg every 3-4 hours. Its intravenous preparation should not be greater than 10% glycerol in 0.4 normal saline as higher concentration results in hemolysis. The complications and toxicity of glycerol therapy include hemolysis, hemoglobinuria, renal failure and hyperosmolor coma.21 Mathew et al reported significant benefit with glycerol in the treatment of acute cerebral infarction.22 The beneficial effect of glycerol was not confirmed in a double blind trial.23 Yu et al in 1992 undertook a double blind stratified and randomized placebo-controlled trial in patients with intracerebral hemorrhage. 107 patients received glycerol (500 ml of 10% glycerol in saline by intravenous infusion over 4 h on 6 consecutive days), and 109 were given saline treatment. At follow-up, all measures of outcome in the treated and untreated groups were similar. At 6 months, mortality rates were 37 in the treated and 33 in the untreated group and improvement in Barthel Index Ratings was not significantly different. Hemolysis was the only adverse effect noted with glycerol. In the absence of a clinically or statistically significant difference in outcome between the treated and untreated groups, this trial provided no justification for glycerol therapy in ICH.24 Hypertonic saline:In 1919, Weed and Mc Kibben reported immediate shrinkage of brain parenchyma on gross visualization after intravenous injection of 30% saline solution in anaesthetized cats. Maximum shrinkage was observed 15-20 min after completion of injection.25 Wilson et al in 1951 reported the effect of various hypertonic salt solutions on cisternal pressure in a group of dogs. The authors evaluated the effect of iso-osmolar doses of 1 M NaCl (5.8%), 1 M Na Lactate (11.2%) and 2/3 M Na succinate (18%) on ICP. The ICP decreased by approximately 10 cm of H2O after administration of hypertonic solutions. The cisternal pressure remained low for 2.5 to 4 hours.26 Besides its osmotic effect, hypertonic saline also improved regional cerebral blood flow.27,28 Hypertonic saline restores normal resting membrane potential by normalizing intracellular concentration of Na and Cl.29 Theoretically, membrane stabilization may help in preserving blood brain barrier; however, this remains unproven.30 The potential complications of hypertonic saline are both neurological and systemic. Abrupt changes in serum osmolality and sodium concentration may result in coma and seizures.31 Both subdural and intracerebral hemorrhages have been observed after abrupt changes in serum sodium.32 Rapid correction of preexisting hyponatremia has been linked to central pontine myelinolysis.33 Prolonged or repeated administration can also result in rebound phenomenon.34 Rapid volume expansion may precipitate congestive cardiac failure in patients with cardiac dysfunction.35 Hypokalemia and hypochloremic acidemia can occur when large amounts of sodium chloride are infused without concomitant potassium replacement.36 The prolongation of prothrombin and activated partial thromboplastin time with decreased platelet aggregation can lead to bleeding complication.37 This is seen when hypertonic saline replaces normal plasma. It also results in phlebitis38 and renal failure.39 Quereshi et al compared equiosmolar dosage of mannintol, 3% NaCl and 23.4% NaCL in a canine model of intracerebral hemorrhage. ICP reduction was most prominent after 23.4% NaCl administration. After 2 hours of administration, only 3% NaCl had a sustained effect on ICP. The ICP in the mannitol group exceeded the pretreatment level. The cerebral perfusion pressure at 2 hours was significantly higher in the 3% NaCl treated group compared to mannitol. The water content in the affected white matter after 2 hrs was also lowest in the 3% NaCl group. The presence of a focal lesion and relative preservation of blood brain barrier may account for the favorable effect of hypertonic saline.40 In a prospective study, Quershi et al did not find any beneficial effect of continuous infusion of 3% saline acetate in 14 patients who had suffered massive stroke.41 Schwartz et al evaluated the effect of hypertonic saline in 6 stroke patients with increased intracranial pressure in whom mannitol was ineffective.42 The maximum ICP reduction after 35 minutes of infusion was 9.9 mm of Hg; thereafter ICP began to rise. Cerebral perfusion pressure was increased although there was no effect on mean arterial blood pressure. They concluded that 75 ml of 10% hypertonic saline reduces ICP and increases cerebral perfusion pressure in stroke patients in whom mannitol was ineffective. All human studies on hypertonic saline for the treatment of cerebral edema and elevated ICP, are based on case reports, case series and small controlled groups. There is no large randomized double blind placebo controlled study on the use of hypertonic saline. A uniform concentration of hypertonic saline has not been studied, dose response curve is lacking and its safety and efficacy need further evaluation. Mannitol:Mannitol has a molecular weight of 182 Dalton and was introduced in neurological practice by Wise and Chater in 1962.43 It is relatively cleared from CSF and brain because of its higher molecular weight, which reduces the rebound phenomenon. Rebound however has been noted in clinical practice.44 Rebound occurs when large dose of mannitol is given at a frequency that exceeds its urinary exertion rate. If sustained hyperosmolality of the brain is obtained, rebound is likely to occur when the plasma osmolality falls more rapidly than the brain. Mannitol in a dose of 1 gm/kg of body weight, given intravenously over a period of 10 to 15 minutes results in raised serum osmolality of approximately 20 to 30 mosm/L, which returns to normal level in about 3 hrs. A dose of 1.5 to 2 gm/kg lowers CSF pressure significantly for 3 to 8 hrs. Lower dosage of mannitol (0.5 gm/kg or less) is desirable to avoid excessive hyperosmolarity and rebound.44 Miller and Leech reported 8 patients studied before and after the administration of mannitol (0.5 gm/kg). The ventricular CSF pressure was reduced in all 8 patients; the maximum reduction (35%) occurred 15 min after administration and it persisted for 45 min. The pressure volume response tested by injecting 1 ml saline in one second into the ventricular fluid showed maximum reduction at 15 minutes. It was concluded that not only mannitol influenced ICP, but also intracranial compliance. Thus an improvement in the neurological status may occur following osmotherapy in spite of little measurable effect on ICP.45 Rheological effects of mannitol Intravenous infusion of mannitol causes influx of extravascular water into the circulation. This leads to acute expansion of plasma volume and increase in cerebral blood flow. This is counterbalanced by compensatory vasoconstriction leading to reduction in CBF, which also results in reduction of ICP. Mannitol results in 15% shrinkage of RBCs, improves deformation and cell wall flexibility thereby improves tissue oxygenation.46,47 This may be important for marginally perfused and hypoxic tissue. The side effects of mannitol are related to its mechanisms of action as osmotic diuretic and by intra vascular volume expansion. Water may move back into the extravascular space, but much will be lost via renal excretion. Thus hemodilution and positive effect on the viscosity of blood following mannitol are lost as the circulating blood volume decreases. This effect may not be obvious after a single dose of mannitol in the newly resuscitated and over hydrated patient, but as dehydration occurs, mannitol becomes less effective, later associated with rebound. Therefore, the diuresed fluid after mannitol administration, must be replaced ml for ml and the smallest effective dose of mannitol should be used. Inadequate fluid replacement may result in renal tubular toxicity. This toxicity is reversible but when high dose of mannitol is combined with vasopressor, aminoglycosides or other nephrotoxic drugs, especially in the presence of hypoxic or hypotensive episodes, devastating nephrotoxicity may occur. Controversies regarding mannitol therapy Hematoma or large hemispheric infarction with surrounding edema act as space occupying lesion producing mass effect and brain herniation. Compartmentalized ICP difference is an important cause of neurological deterioration rather than a global increase in ICP.48,49 Every hypothetical mechanism of action of mannitol has a maximal effect in the normal hemisphere which can lead to potential aggravation of dangerous pressure difference because of differential lowering of compartmentalized ICP. It has been suggested that mannitol may aggravate midline shift and lead to neurological deterioration.50 Garcia et al in their experimental study found that a high dose of mannitol led to 21% ICP reduction over the injured hemisphere and 26% reduction over the normal hemisphere.51 Videen et al studied 6 patients who had acute middle cerebral arterial infarction and CT evidence of midline shift measured using the brain boundary shift on sequential T1weighted images acquired before and after a 1.5 gm/kg bolus infusion of mannitol. At 50 to 55 minutes after the baseline scan, total brain volume significantly decreased. The non-infarcted hemisphere shrank more compared to the infarcted hemisphere. However, the clinical implication of this study could not be ascertained.52 Kauffman et al in an experimental study investigated the pharmacokinetics of mannitol administered for treatment of vasogenic cerebral edema. Control animals received no mannitol, while the treated group received either a single dose or five doses of 0.33 gm /kg of radiolabeled mannitol administered 4-hourly. Water content measurement showed that a single dose of mannitol failed to reduce cerebral water content or edema progression at 4 hrs, while multiple doses produced 3% increase in water content in edematous region. It was concluded that reversal of the osmotic concentration gradient between edematous brain and plasma develops following multiple mannitol injections, which was associated with exacerbation of vasogenic cerebral edema.53 Manno et al evaluated the effect of a single large dose of mannitol on midline shift after large cerebral infarction in seven patients. The final average change in midline shift compared to the initial displacement was only 0.0 + 1 mm in horizontal and 0.25 + l.3 mm in vertical. Stroke scale improved in two, Glasgow coma scale score improved in three and pupillary light reactivity returned in two patients. No patient worsened. They concluded that a single large dose of mannitol used in patients with infarction did not alter midline shift or worsen neurological status.54 Similarly, Paczynski et al, demonstrated in the rat model that repeated mannitol infusion reduced the water content of both edematous and normal brain tissue in contrast to earlier studies. Though midline shift was not directly measured in this model, the mean difference in water content between hemispheres was small when mannintol was used in a dose of 0.33 gm/kg. Large and repeated dosing of mannitol however, led to progressive increase in water loss from the normal hemisphere.55 Despite the fact that mannitol has been widely used to decrease elevated ICP in both ischemic and hemorrhagic strokes, randomized clinical trials are very few. Most of the trials were confounded and a randomized clinical trial of mannitol in intracerebral hemorrhage is lacking.56 Wang studied 44 cases of acute hemorrhagic stroke treated with FCMCK therapy and compared them with 44 cases treated with mannitol. The mortality rate of the FCMCK treated group was 4.5% which was significantly lower than that of the mannitol group (47.7%).57 Gigliuto et al reviewed 20 patients with ICH admitted to intensive care units with raised ICP. They concluded that there was no significant therapeutic benefit using mannitol osmotherapy.58 Based on these small clinical and experimental trials, no conclusion can be drawn. The American Heart Association, in their guidelines for the management of spontaneous intracerebral heamorrhage, recommend mannitol 20% (0.25 gm/kg every four hours) in patients with type B ICP waves, progressively increasing ICP and clinical deterioration due to mass effect.59 However, enough evidence is lacking for the routine use of mannitol in ICH. Conclusion Over the years, some of the treatment options for raised ICP such as barbiturates or hyperventilation have lost their significance. Hypertonic solutions such as urea, hypertonic saline, glycerol and mannitol have been used to lower ICP for a long time. A large number of clinical and experimental studies demonstrated that a single dose of mannitol reduces elevated ICP transiently. The enthusiasm about mannitol for patients with ICH however is dampened by several factors: 1) Most of the clinical studies on mannitol are on patients with head injuries; 2) No randomized study on ICH patients has been undertaken, and the long-term beneficial effect of mannitol is unknown; and 3) There is evidence that repeated infusion of mannitol may even aggravate brain edema. Hypertonic saline infusion has been reported to reduce ICP when mannitol fails. However, this observation is based on small series and case reports. A properly designed randomized placebo controlled study is therefore needed to evaluate the role of mannitol and hypertonic saline in different concentrations, to address this issue. References
Copyright 2003 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni03031f1.jpg] [ni03031f2.jpg] |
| |||||||||

{kind=link}
{kind=link}