 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
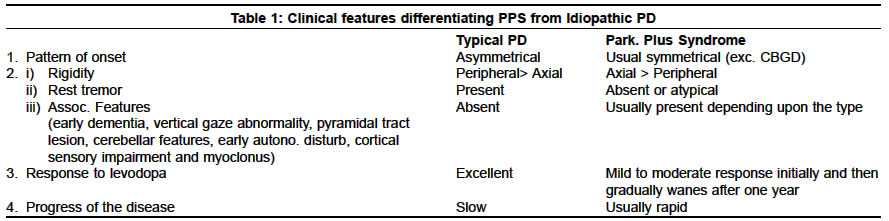
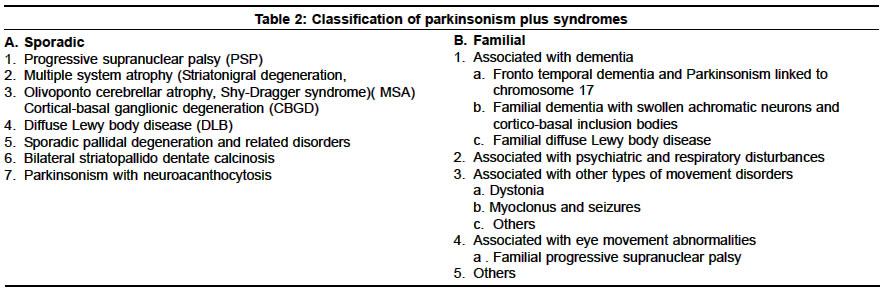
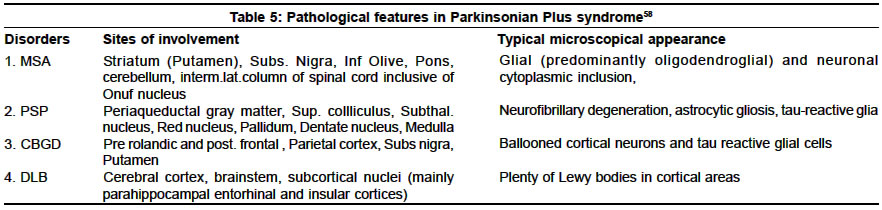
Neurology India, Vol. 51, No. 2, April-June, 2003, pp. 183-188 Parkinsonism plus syndrome - A review K. Mitra, P. K. Gangopadhaya, S. K. Das Shyamal Kumar Das Accepted on 10.08.2002. Code Number: ni03059 ABSTRACT Parkinsonism plus syndrome is a group of heterogeneous degenerative neurological disorders, which differ from the classical idiopathic Parkinson's disease in certain associated clinical features, poor response to levodopa, distinctive pathological characteristics and poor prognosis. Associated clinical features include symmetrical onset, infrequent or atypical tremor, prominent rigidity in axial musculature, bradykinesia, early postural instability, supranuclear gaze palsy, early autonomic failure, pyramidal affection, cerebellar involvement, alien limb phenomenon, apraxia and significant early cognitive dysfunction in some cases. Progressive supranuclear palsy (PSP), multiple system atrophy (MSA) and dementia with Lewy body disease (DLB) are commoner disorders. Less frequent disorders are cortico-basal ganglionic degeneration (CBGD), frontotemporal dementia with chromosome 17 (FTDP-17), Pick's disease, parkinsonian-dementia complex of Guam, Pallidonigral degeneration, Wilson's disease and a rigid variant of Huntington's disease. During the last 3 decades, major progress has been made in understanding PSP, CBGD and FTDP-17, which are tau disorders. MSA and DLB together with idiopathic Parkinson's disease are called α-synucleinopathies. Recent studies show that the diagnosis of these Parkinsonism plus syndromes improves when strict diagnostic criteria are used. However, unusual presentations may pose a diagnostic challenge. The shortcomings of the current studies demand the need for further research to identify biologic markers that may allow earlier diagnosis, and understanding of the factors leading to α-synuclein or tau aggregation. Identification of therapeutic strategies that may prevent the aggregation of these proteins and rescue dysfunctional cells has been stressed. This review focuses on the advances in the clinical, neuroimaging, pathologic, genetic and management aspects of these disorders. Key Words: Parkinsonism plus syndromes, Progressive supranuclear palsy, Multiple system atrophy, Cortico-basal ganglionic degeneration, Dementia with Lewy body disease. Idiopathic Parkinson's disease (IPD) is characterized by rigidity, bradykinesia, rest tremor, impaired postural reflexes, asymmetric onset, good to excellent response to levodopa, and pathologically by degeneration of pigmented neurons in substantia nigra along with the presence of Lewy bodies. Approximately 80-85% of Parkinsonism patients seen in movement disorders clinics are found to have typical features of IPD; the remaining 15-20% patients belong to the other category of neurodegenerative disorders. Less commonly, Parkinsonism secondary to drugs or toxins, brain tumors, hydrocephalus, multiple infarcts and viral infection is observed. A great emphasis is being placed on the identification of clinical clues in patients who have a clinical diagnosis of IPD with additional or atypical clinical features. These are classified as having Parkinsonism plus syndromes (PPS).1 Several differences exist between typical IPD and PPS in the pattern of extrapyramidal features, pattern of onset of illness, and therapeutic response—these have been given in Table 1.2 A positive family history associated with a number of PPS cases suggests that sporadic and familial classification of PPS is necessary (Table 2).3 In this review, we present the recent concept on classification, clinical features, treatment, neuroimaging findings, pathological features, and the advances of molecular genetics among the commoner varieties of PPS. Familial Parkinsonism Plus Syndromes There is phenotypic heterogeneity in these kindred: Parkinsonism plus dementia or dementia alone. Additional clinical features include disinhibition, apathy, poor impulse control, psychosis, alcoholism, dystonia, eye movement abnormalities such as vertical gaze palsy, eyelid opening and closing apraxia, upper and lower motor neuron dysfunction. Similarly, the neuropathological findings show some heterogeneity. Tau-positive cytoplasmic inclusion or widely distributed ballooned neurons are present in some of the kindred while absent in others. The pathological features are similar to PSP in some, and to CBGD in others.6 The available genetic evidence linking the phenotype to a region on the long arm of chromosome 17 suggests that they may represent one disorder and that phenotypic heterogeneity may represent different mutations of the same gene. Two unrelated English kindred were reported in which affected family members presented with personality changes, dementia, Parkinsonism, limb clumsiness and disequilibrium. Disease progression was rapid, leading to death in 3 to 13 years. At autopsy, neuronal loss and gliosis with swollen achromatic neurons in the cortex and corticobasal inclusion bodies in the substantia nigra were noted. These two kindred might represent familial CBGD.7 There are no published reports linking familial DLB to a particular chromosomal region. Six kindred with affected individuals displayed a combination of Parkinsonism, depression, hypoventilation and weight loss.8 Resting tremor and bradykinesia were seen in 3 of them; levodopa responsiveness was seen in all kindred initially, but Lewy bodies were present in only 2.9 The molecular genetic status of these individuals is unknown. Kindred presenting with X-linked dystonia and Parkinsonism (i.e. Lubag disease) demonstrated all cardinal features of Parkinsonism, including a positive response to levodopa.10 The genetic defect has been linked to a region in the X chromosome.11 The phenotype of kindred with dopa-responsive dystonia has been linked to genetic markers on the long arm of chromosome 14.12 There have been published reports of familial PSP.13,14 Affected patients show Parkinsonism with eye movement abnormalities. The molecular genetic status of these kindred is unknown. Myoclonus and seizures are part of the clinical spectrum of the affected Spellman-Meunter family.15,16 Young onset, rapidly progressive akinetic rigid Parkinsonism, characterizes this kindred. The disorder is poorly responsive to levodopa and includes ataxia, profound dementia, dysarthria and rapid weight loss. On autopsy, cortical and subcortical Lewy bodies were seen. In several other Parkinsonian kindred, myoclonus, seizures or both are present or both are part of the phenotype.17 Olfactory dysfunction is a phenotypic characteristic of familial Parkinsonism, independent of kindred phenotype.18 Sporadic Parkinsonism Plus Syndromes Recently, based on histopathological features, the Parkinsonism syndromes have been classified into 2 groups, "taupathies" and "α-synucleinopathies".21 In both groups, there are abnormal filamentous inclusions in neurons and glial cells.22 Abnormal phosphorylated tau proteins are a basic component of neuronal and glial cell inclusion in PSP and CBGD, hence they are regarded as taupathies.23,24 Abnormal phosphorylated tau proteins constitute many pathological hallmarks which are known as neurofibrillary tangles, neuropil thread, Pick bodies, and granulo-vacuolar degeneration. Alzheimer's disease, Pick's disease and FTDP-17 are also grouped in this category. α-synuclein is an important component of Lewy body in IPD and DLB. α-synuclein disturbance also takes part in the formation of glial cell inclusion in MSA. These disorders are called "α-synucleinopathies."25-27 Recent advances in neuroimaging have helped in differentiating IPD from PPS. PET has documented lower posterior putamen / caudate uptake ratio of 18F-dopa in IPD than in PSP and control.28 3D MRI-based volumetry can distinguish atypical Parkinsonism from IPD.29 Recently, SPECT study has helped in imaging the pre and post-synaptic dopaminergic pathways by using different ligands.30 Progressive Supranuclear Palsy (PSP) PSP is a diagnosis based on clinical criteria (Table 3) and there are no specific laboratory tests to confirm it.32 In CT and MRI scan, there is atrophy of the dorsal mesencephalon and a widening of the aqueduct. In MRI, periaqueductal hyperintensity in T2-weighted images is noted. This finding is probably secondary to gliosis in the later stages of the illness.34 PET scan, using 18FDG, shows hypometabolism in the frontal cortex and flurodopa PET shows decreased labeling in both the putamen and the caudate nucleus.35 There is decreased saccadic amplitude by electrooculography in PSP. Barium swallow study shows barium residues on the base of the tongue, vallecula and upper esophagus in PSP. 36,37 There is no specific drug therapy. In the early stages, mild to moderate improvement in symptoms may occur with levodopa or dopamine agonists, but improvement fails to be maintained later because of the loss of D2 receptors due to the degeneration of post-synaptic striatal neurons. Symptomatic therapy helps in walking difficulty, eating problem, dysarthria, blepharospasm, depression, and excessive drooling. Botulinum toxin has been used in cases with prominent levator lid inhibition and is quite beneficial. The etiopathogenesis of PSP remains unknown and only a few studies have evaluated the presence of a genetic predisposition. Two studies confirmed that the distribution of apolipoprotein E E4 alleles frequencies in PSP patients was similar to that in control individuals. They provided substantial evidence of a lack of association between apo E and the development of PSP.33 Several authors found that a dinucleotide repeat polymorphism in a tau intron was more frequently found in PSP patients than in a group of control individuals. The high frequency of this allele expression in the general population excludes it as a marker for PSP. Whether predisposing genetic factors lead to an abnormal tau phosporylation and / or induce abnormal mitochondrial ATP production needs to be explored. Similarly, whether cytokine production by active astrocytes and microglia may contribute to this process needs to be investigated. Multiple System Atrophy (MSA) MSA is a sporadic disease comprising striato-nigral degeneration (SND), Shy Drager syndrome (SDS) and many cases of sporadic olivopontocerebellar atrophy (OPCA). Clinical features of MSA have been summarized in Table 4. It is now recognized that dysautonomia (of varying degree) is a nearly universal feature of patients with MSA. Recently, new terms such as MSA-P and MSA-C have been proposed. Patients with MSA often present with a predominantly Parkinsonism form of the disorder (striato-nigral variety called MSA-P) or a largely cerebellar form (OPCA-variety called MSA-C). Although most patients demonstrate one of these two forms, many have overlapping features, often with pyramidal tract deficit in addition to dysautonomic features.38,39 Erectile dysfunction may be the first symptom in men with MSA, predating the onset of any other neurological symptom by several years. Nothing is known about the sexual dysfunction of women with MSA. Heavily T2 - weighted MRI images shows low intensity signal in the putamen (possibly caused by excessive iron deposition) equal to or greater than that normally present in the globus pallidus along with an infrequent linear hyperintensity lateral to the putamen. In those with MSA-C, there may be cerebellar and pontine atrophy. 18F-DOPA cannot reliably distinguish between MSA and IPD. As a group, patients with SND show lower striatal glucose metabolism than those with IPD. Phosphorus MR spectroscopy may disclose differential contents of phosphate metabolite and ion content in both MSA and IPD which may help to differentiate MSA from IPD.40 Patients with MSA show reduced phospho-creatine, increased phosphate and unchanged cytosolic free magnesium ion, whereas IPD patients show unchanged phospho-creatine, increased phosphate and decreased free magnesium ion.41 In MSA, anorectal function study shows an abnormal staining pattern, decreased anal tone or dysfunction of both which are indicative of Onuf nucleus dysfunction.. EMG study of anal sphincter may show denervation. It usually occurs earlier and develops faster than in IPD.42 Motor-evoked potential is normal in MSA, but somatosensory-evoked potential abnormalities are found in 40% of MSA with no significant differences between MSA-C and MSA-P subgroups. Abnormal latencies of wave III in BAER are more frequent in MSA-C indicating brainstem dysfunction.43 The pathological changes in MSA consist of cell loss and gliosis in striatum (mainly putamen) and substantia nigra, inferior olives, pons and cerebellum, in the intermediolateral cell columns, and Onuf nucleus in the spinal cord. Oligodendroglial cytoplasmic inclusions are characteristically present, widely and in plenty, but Lewy bodies are usually absent, unless incidental.38,39 The management of patients with MSA is usually complex. Parkinsonism may respond markedly to levodopa in the early stages, but the response is usually incomplete and short-lived. Amantadine, by its dopamine agonist effect, may be helpful in early cases. Symptomatic treatment of orthostatic hypotension, urinary incontinence or retention, impotency and bowel dysfunction should be taken care of. Botulinum toxin may be helpful for antecollis. Tracheotomy or anchoring one vocal cord in abduction or a laser crico-arytenoidectomy has been considered for respiratory stridor. A recent suggestion has been made about the role of environmental toxins in MSA pathogenesis.26 Cortico-basal Ganglionic Degeneration (CBGD) Depression and obsessive-compulsive neurosis are common. CT and MRI scan shows cerebral atrophy contralateral to the clinically affected side (typically, the right hemisphere). PET study reveals asymmetric reduction of cortical FDG metabolism and striatal tracer uptake (18F-dopa).47 Neuropsychological testing shows greater right hemispheric dysfunction.48,49 Electro-oculography reveals abnormal eye movement in the horizontal plane characterized by increased latency of saccades, perhaps due to parietal cortex involvement in CBGD. Pathologically, there is asymmetrical cerebral atrophy involving pre-frontal and parietal regions.46 Depigmentation of substantia nigra and degeneration of putamen and striatum is noted. A recent study revealed ballooned neurons, immunoreactive with anti-B crystalline, a sensitive marker of swollen neuron in CBGD.50 Based on the relative distribution of pathology, 2 types of presentation have been documented—one is anterior cortical predominance, which manifests as frontotemporal dementia (morphologic similar to Pick's disease51) and another is lateral dominant hemisphere frontal lobe which presents as primary progressive aphasia. The course is gradually progressive. The disease is notoriously resistant to symptomatic therapy. Clonazepam is useful in myoclonus. Baclofen and botulinum toxin has been tried for dystonia and pain. Late onset dysphasia is associated with poor survival. Diffuse Lewy Body Disease (DLB) The clinical picture of established cases of DLB is that of a particular dementia syndrome with features of Parkinsonism. Cognitive deficits may emerge in the course of Parkinson's disease, but the presenting feature is usually the cognitive impairment with later emergence of Parkinsonism. The key clinical features are fluctuation and visual hallucination. Another diagnostic clue is marked neuroleptic sensitivity. Some patients have prominent sleep disturbance and others have falls and syncopal episodes. Rarely, individuals may have prominent myoclonus and rapid deterioration, which may suggest Creutzfeldt-Jakob disease.50 Structural neuroimaging often shows atrophy and commonly additional white matter changes, often attributed to ischemia. Few studies of functional imaging have been done, although some authors demonstrated a temporo-parietal pattern, along with more posterior changes including to the occipital cortex (this may be relevant to the hallucination). EEG usually shows diffuse slowing. The pathologic hallmark is the Lewy body, plenty in the cortex in addition to basal ganglia, best demonstrated with anti-ubiquitin immunostaining. A major component of the Lewy body has been demonstrated to be α-synuclein. In association with Lewy bodies, the neurons show neuritic change, demonstrated both with ubiquitin and synuclein immunostaining, known as Lewy neurite.52-54 Management is difficult. Anti-Parkinsonian medication may help in alleviating akinesia but can increase hallucination and confusion. Conversely, dopamine-blocking neuroleptics can markedly worsen the extrapyramidal syndrome and cognitive function. If the motor symptoms are prominent, these are best managed with small doses of L-dopa; anticholinergic drugs should be avoided. Similarly, neuroleptics should be avoided, although some evidence exists that the newer atypical neuroleptics such as quetiapine may be safer. Sporadic Pallidal Degeneration Since the etiology of these disorders is unknown, no specific or causal treatment is available and only early genetic counseling of the population at risk may be effective in reducing subsequent incidence of these extremely rare diseases. Symptomatic treatment may include physiotherapy, and the use of L-dopa on patients with prominent akinetic-rigid Parkinsonism, while antispastic drugs and neuroleptics may be of use in those individuals with prominent pyramidal and hyperkinetic clinical features. Bilateral Striatopallidodentate Calcinosis Parkinsonism with Neuroacanthocytosis Pathological features in Parkinsonian plus syndrome are summarised in table 6. CONCLUSION Patients with Parkinsonism-plus syndromes represent a relatively small portion of Parkinsonism patients seen in general and movement disorders clinics. Given the wide spectrum of disease phenotypes associated with these disorders and the often subtle clinical differences, establishing the correct diagnosis, especially at disease onset, can be difficult despite serial clinical observations and repeated neurological examination. Autopsy confirmation is often necessary. Advancement in molecular genetics may provide a better understanding of some of these rare syndromes and holds substantial promise for more rational classification and therapy. Moreover, an accurate diagnosis of these disorders is necessary to understand their cause and pathogenesis. This may allow development of biologic therapeutic strategies to stop or slow disease progression, such as inhibition of tau or α-synuclein aggregation. ACKNOWLEDGEMENTS We gratefully acknowledge the help of Prof Oksana Suchowersky, Director of Movement Disorders Clinic, Univ. of Calgary, Canada for reviewing the article and providing useful suggestions. We are also thankful to Miss Aparna Dutta for secretarial assistance. REFERENCES 1. Jankovic J. Parkinsonism - plus Syndromes. Mov Disord 1989;4:95-119
Copyright 2003 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni03059t3.jpg] [ni03059t1.jpg] [ni03059t4.jpg] [ni03059t5.jpg] [ni03059t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}