 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
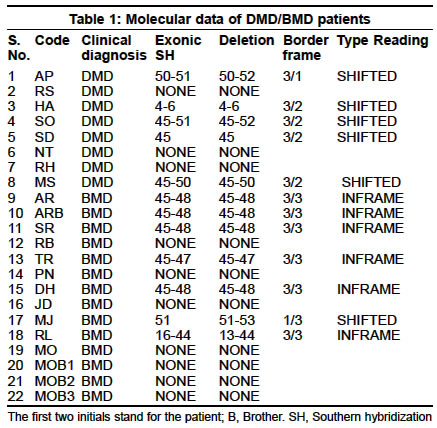
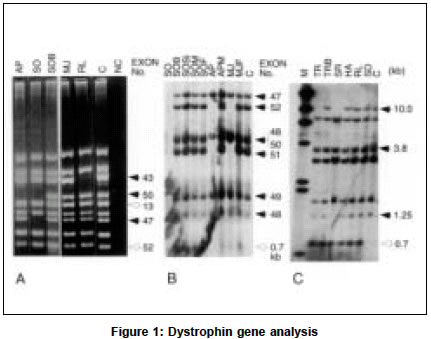
Neurology India, Vol. 51, No. 2, April-June, 2003, pp. 223-226 Deletion analysis of the dystrophin gene in Duchenne and Becker muscular dystrophy patients: Use in carrier diagnosis D. Kumari, A. Mital, M. Gupta,* S. Goyle Human Molecular Genetics Laboratory, School of Life Sciences, Jawaharlal Nehru University, New Delhi, India, *Department of Neurology, G. B. Pant Hospital, New Delhi, India. Daman Kumari Accepted on 21.06.2001. Code Number: ni03069 ABSTRACT The dystrophin gene was analyzed in 8 Duchenne muscular dystrophy (DMD) and 10 Becker muscular dystrophy (BMD) unrelated families (22 subjects: 18 index cases and 4 sibs) for the presence of deletions by multiplex polymerase chain reaction (mPCR; 27 exons) and Southern hybridization using 8 cDMD probes. Deletions were identified in 5 DMD and 7 BMD patients (6 index cases and 1 sib). The concordance between the clinical phenotype and "reading frame hypothesis" was observed in 11/12 patients (92%). The female relatives of DMD/BMD patients with identifiable deletions were examined by quantitative mPCR. Carriers were identified in 7 families. We also describe a variation in the HindIII pattern with cDNA probe 8 and 11-14. Molecular characterization of the dystrophin gene in this study has been helpful in advising the patients concerning the inheritance of the condition, and carrier diagnosis of female relatives, and should also prove useful for prenatal diagnosis. Key Words: Carrier diagnosis, Dystrophin, Deletions, Multiplex PCR. INTRODUCTION Duchenne and Becker muscular dystrophies (DMD/BMD) are allelic, X-linked, recessive neuromuscular disorders caused by mutations in the dystrophin gene located at Xp21.1. Due to the extremely large size of the gene (2.4 Mb), majority of the mutations are deletions (65%) or duplications (5%).2 The deletions in the dystrophin gene are non-randomly distributed in two regions, at the 5' terminus (minor "hotspot") and in the distal half of the central rod domain around exons 44-53 (major "hotspot").2,3 Mutations that disrupt the open reading frame (ORF) cause DMD, whereas those which maintain it result in BMD.4 Overall, about 92% of deletions conform to the "reading frame rule".5 Although the dystrophin gene has been analyzed extensively all over the world, only a few studies have been reported on Indian patients.6-8 These reports were either confined to deletion analysis by mPCR alone7 or combined with Southern hybridization with cDNA probes limited to the hotspot region.8 A complete restriction analysis of the gene is important because a few deletions have also been mapped to the 5' and 3' regions of the dystrophin gene. We, for the first time, reported an analysis of the entire dystrophin gene by Southern hybridization with 8 cDNA probes in Indian DMD patients.9 The present study has been extended to Indian BMD patients and their family members. Our study of 18 unrelated DMD/BMD families describes gene deletions for which precise exon boundaries were determined by Southern blot analysis. The concordance between the clinical phenotype and reading frame hypothesis was evaluated for this patient population. Furthermore, we report the diagnosis of carrier status in the female relatives of the patients with deletions, the presence of an extra HindIII fragment with cDMD probe 8 and confirmation of the variation in the HindIII restriction pattern with cDMD probe 11-14. MATERIAL AND METHODS Patients were diagnosed at the Department of Neurology, G. B. Pant Hospital, New Delhi and referred to our laboratory for DNA analysis. The diagnosis was established by clinical assessment, family history, grossly raised serum creatine kinase (CK) and typical muscle histology or electromyography or both. DMD cases were usually differentiated from BMD by the age at which the patient became wheelchair-bound. All index cases had calf hypertrophy and markedly raised serum CK levels. A total of 46 heparinised peripheral blood samples were obtained from 18 unrelated DMD and BMD families with prior informed consent. Of these, 8 were DMD and 14 BMD patients, 7 sibs (4 males, 3 females), 1 nephew and 16 parents (10 mothers and 6 fathers). Two cases of DMD were chair-bound by 7 and 10 years of age, and 6 below the age of 11 years, although ambulatory, walked with difficulty. The age of the loss of ambulation in 2 BMD sibs (MOB1 and MOB2) was around 32 years while the others were ambulatory (age range 9-43 years). Two DMD (RS, HA) and four BMD patients (AR, PN, DH, MO) had a positive family history. The control blood samples were from 20 unrelated normal healthy individuals (11 male and 9 female) who had no family history of any muscle disease. Genomic DNA was isolated from peripheral, essentially as described elsewhere.9 The frequency and distribution of deletions in the dystrophin gene were assessed by PCR using three different multiplex primer sets comprising 9 pairs each (IMMCO, U.S.A.) as described earlier.9 The deletions were confirmed in doubtful cases by repeating the amplification and electrophoresis. Normal male DNA served as a positive control and reaction without template DNA as a negative control in each set of the PCR reactions. Southern hybridization of HindIII (New England Biolabs, U.K.) digests using 8 cDMD probes (American Type Culture Collection, U.S.A.) was carried out as described previously.9 Peripheral blood was available from the female members in 4/11 families with exonic deletions. In addition, peripheral blood was obtained from 8 female relatives of 5 DMD patients (JI, PD, FZ, CH, UR).9 QM-PCR was carried out as mPCR except that 5µci of að-32P-dCTP (BRIT, INDIA) was included in each reaction along with the cold nucleotides and 4-5 pairs of primers. The multiplex set was specially made using primers for both deleted and intact exons. The PCR cycle was limited to 18 cycles. Eight microliters of the reaction products were separated on 2.5% agarose. After electrophoresis, the gel was fixed and dried as described earlier,10 and exposed to Hyperfilm MP (Amersham Pharmacia Biotech, U.K.) overnight. The quantitative analysis was done using Collage software (Photodyne Inc., U.S.A.). Background determination was done manually. After automated integration, individual peak areas were expressed as percentages of the total area of the peaks in each sample. Normal male DNA was always included as control in each set of the PCR reactions. RESULTS The dystrophin gene was analyzed in DMD/BMD patients by mPCR and Southern hybridization. Deletions were detected in 5/8 DMD patients (62.5%), 6/10 BMD patients (60%), and in 1 symptomatic sib (Table 1). Eleven deletions were localized to the major hotspot region between exons 44 and 52. Only 1 deletion was detected at the 5' end of the gene (HA). The clinical severity of the disorder in patients with deletions was tested against the "reading frame hypothesis". The border type and the effect of deletion on the translational reading frame are given in Table 1. Deletions that created a frame-shift mutation in the protein coding region caused DMD, whereas inframe deletions caused BMD. One patient (MJ) did not conform to the hypothesis. A deletion of exon 51 was identified in MJ by PCR which extended to include exons 52 and 53 by Southern analysis. Both were frame-shift deletions. The patient was 10 years old, ambulatory with slight difficulty in walking. Although the symptoms began at age 2 (typical of DMD) the clinical course was mild which is characteristic of BMD. In 4 patients, the deletion pattern observed by the Southern blot analysis was different from that observed by PCR. Exon 52 was amplified by PCR in 3 patients (MJ, AP and SO, Figure 1a) but was found deleted on Southern blot analysis (Figure 1b). In another patient RL, deletion of exons 13-44 was observed by Southern analysis but intermittent exons 13, 25 and 42 were amplified by PCR. Southern analysis with Pst I also revealed the deletion of exon 52 in AP and SO (data not shown). The DNA sample of RL and MJ was insufficient for digestion with another restriction enzyme. During the course of this study of deletion analysis in DMD/BMD patients and their male sibs by Southern hybridization, an extra 0.7 kb HindIII fragment was observed in 7/8 DMD and 9/14 BMD patients with cDNA probe 8 (Figures 1b & 1c). This fragment was absent in the unrelated control in the same blot. The Southern hybridization was extended to all the male and female members of the probands. The 0.7 kb extra fragment was observed in 16/18 families. One exception was the absence of this fragment in a DMD patient (AP) while it was present in his mother. The blood samples from the other members of this family were not available. Probe 11-14 identified a total of 10 bands (10.0, 7.8, 6.8, 6.0, 5.9, 2.8, 2.1, 1.9/1.8, 1.5 and 1.45 kb). A strongly hybridizing fragment of 2.8 kb was present in each of the subjects, while 1.9 and 1.8 kb bands appeared as a single, faint, co-migrating fragment (data not shown). We had previously reported this polymorphism in Indian DMD patients.9 We also analyzed the pattern in more Indian individuals as well as in individuals of other ethnic origins11 and concluded that the 2.8 kb fragment seen with probe 11-14, which was not observed by Darras and Francke,12 belongs to the dystrophin gene. The carrier status of the female relatives of the DMD/BMD patients was determined by QM-PCR in 1 BMD and 8 DMD families with identifiable deletions. A total of 9 mothers, 4 sisters and 11 normal males (7 fathers, 2 brothers and 2 controls) were examined. Figure 2 illustrates the results of QM-PCR in 1 representative family (JI). JI and his brother (JIB) had deletion of exons 47-50. The selected primers used in QM-PCR are for exons 48, 49, 51 and 12. Examination of the relative band intensity of the father (JIF) and the mother (JIM) indicated that the mother had reduced relative band intensity for the 2 deleted exons (exon 48 and 49) in her son, to about 50% of the level found in the father showing that she is a carrier. One BMD (DHM) and 6 DMD (MSM, APM, JIM, PDM, FZM, URM) mothers, and 2 DMD sisters (PDSi, FZSi1) were carriers of the deletion found in their respective sons/brothers. Two DMD mothers (SOM, CHM) and 2 DMD sisters (SOSi, FZSi2) did not show the expected deletion and hence were not carriers (data not shown). DISCUSSION The partial gene deletions identified in 61% of DMD/BMD patients were distributed non-randomly in two hotspot regions of the dystrophin gene. Their frequency and distribution in the Indian population is similar to other populations.13 However, this is slightly lower than the frequency observed in Indian patients earlier.6,7,9 This difference could be due to the fact that earlier studies were done only with DMD patients9 or they had more DMD patients than BMD patients.6,7 In patients with deletions, the clinical severity of the disorder was examined for the frame-shift hypothesis. The results, with the exception of 1 patient (MJ), agree with other studies.14 He has a frame-shift deletion but the clinical progression of the disease is more like BMD. It has been reported earlier that alternate splicing events can produce an unpredictable coding sequence.15 In MJ, alternate splicing may explain the observed deviation from the frame-shift theory, as splicing out exon 50 would maintain the reading frame. Analysis of the mRNA and dystrophin immunostaining can help resolve this discrepancy. Discrepancies between the results obtained with Southern and mPCR analysis have been reported earlier.16-18 This is attributed to a low resolution of Southern analysis, and sequence variation within priming site that prevents annealing of primer. In the present study, exon 52 was amplified by PCR in 3 patients (AP, SO and MJ) but the 7 kb HindIII fragment containing exon 52 was found to be deleted by Southern hybridization. Analysis with Pst I also showed the deletion of exon 52 in AP and SO. This false amplification has been observed in 7 patients by us in an earlier study9 and has also been reported earlier 18 in 2 patients. Another patient (RL) showed a large deletion, of exons 13-44 by cDNA analysis, but intermittent exons 13, 25 and 42 were amplified by PCR. Further hybridization on different restriction enzyme digest and PCR amplification with another pair of primers, specific for the same region may help to resolve this discrepancy. The 0.7 kb extra HindIII fragment observed with probe 8 in this study has also been seen in 11/13 muscular dystrophy patients and their family members who were screened for dystrophin abnormality (Kumari et al, unpublished data). The appearance of this fragment could not be due to the incomplete digestion of the genomic DNA because it is smaller than the normal fragments identified with cDNA 8 and the intensity is comparable to other bands. Initial studies aimed at understanding the origin of this fragment by probing with labeled exons indicate that this fragment may not be a part of the dystrophin gene (Varma et. al, unpublished data). But why this fragment is not seen in the control samples is not clear. To understand the origin and relevance of the 0.7 kb extra HindIII fragment observed with cDNA probe 8 in DMD/BMD families, more number of controls need to be analyzed. For carrier detection, the mPCR products were analyzed during the exponential phase of PCR, a stage at which the genomic proportions of the different exons are expected to be maintained. It has been possible to detect the deletion of specific exons in probable carriers by a reduction in the relative intensities of the corresponding PCR fragments, to about 50% of the level in normal individuals. Because 30% of the DMD/BMD cases originate de novo, not all mothers are carriers. Carrier diagnosis is thus important for genetic counseling and prenatal diagnosis of DMD/BMD. The use of primers for both the deleted and intact exons in the multiplex set used for QM-PCR helps in the direct comparison of the intensity of the amplified product in the normal and carrier individual. To conclude, an accurate molecular diagnosis achieved in about 61% of DMD/BMD patients has been helpful in advising the patients about the inheritance of the condition and the analysis of the carrier status of the female relatives, and should also prove useful for prenatal diagnosis. ACKNOWLEDGEMENTS This study was supported by the Indian Council for Medical Research, New Delhi. DK and AM were funded by a University Grants Commission fellowship. REFERENCES 1. Hoffman EP, Kunkel LM, Dystrophin abnormalities in Duchenne/Becker muscular dystrophy. Neuron 1989;2:1019-29.
Copyright 2003 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni03069f1.jpg] [ni03069t1.jpg] [ni03069t2.jpg] |
| |||||||||

{kind=link}
{kind=link}