 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 51, No. 2, April-June, 2003, pp. 227-234 Primary degenerative cerebellar ataxias in ethnic Bengalees in West Bengal: some observations A. Chakravarty, S. C. Mukherjee Department of Neurology, Vivekananda Institute of Medical Sciences, Medical College, Kolkata-700006, India.
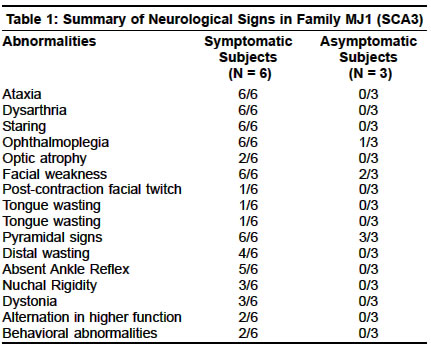
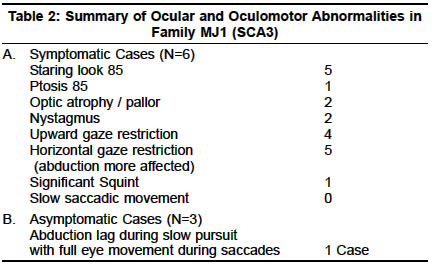
Accepted on 26.05.2001. Code Number: ni03070 ABSTRACT Seventy cases of primary degenerative cerebellar ataxias in ethnic Bengalees from southern West Bengal, India, were studied by the authors. Of these, 50 cases were of the familial type (hereditary ataxias) encountered in 23 families and the remaining 20 were of sporadic onset. 18 cases (from 11 families) were of `probable' autosomal recessive (AR) inheritance, 12 cases (8 families) had Friedreich's type ataxia (FA), 4 cases (2 families) had FA type ataxia with retained reflexes and in 2 cases (1 family) the exact phenotypic characterization could not be made. AR inheritance in these cases seemed most likely in view of the occurrence in a single generation with unaffected parents and history of consanguinity in many of the families studied. Genotypic confirmation of FA type ataxia and its variants could not be done in any case due to the non-availability of technology for studying the FA locus but some common dominant ataxia genotypes could be excluded. Thirty-two cases (from 12 families) with autosomal dominant ataxias (ADCA) were studied. Genotype analysis revealed 4 families with SCA2 genotype, 5 families with SCA3 and 3 families where genotypic characterization could not be made (phenotypically 2 were of ADCA I and 1 of ADCA II). No clear preponderance of one particular genotype of SCA over another could be demonstrated in our ethnic Bengalee patients. We also noted significant intra and inter-family variations in phenotypes within the same genotypic form as well as overlapping of clinical signs between different genotypes. Slow saccades and peripheral neuropathy were not seen consistently in our ethnic Bengalee subjects with SCA2 genotype. Similarly, extrapyramidal features, ophthalmoplegias and distal amyotrophy were seen in some but not all families with the SCA3 genotype. Phenotypic expression appeared to be an inconsistent marker of the SCA genotype in our patients. Of the 20 sporadic cases with cerebellar ataxia, genotype analysis revealed 2 cases with SCA1 and 1 with SCA2. Some of the sporadic ataxia cases had extracerebellar involvement and may warrant classification as Multiple System Atrophy. In all the 3 subjects with genotype characterization, phenotype correlation was lacking. The clinical pattern of hereditary ataxias in ethnic Bengalees seems to be somewhat different from that seen in Western India. The need for clinical and genetic studies of ataxias in different specific ethnic populations of India has been stressed. Key Words: Cerebellar ataxia, Autosomal recessive, Autosomal dominant, Ethnic variations. Diagnosis and classification of primary degenerative cerebellar ataxias remains a challenging problem to clinical neurologists. Advances in molecular genetic technology in the last few years have thrown light on the subject though several issues remain unresolved. Interest in research in the field of hereditary ataxias in India, had been kindled undoubtedly by Wadia and his colleagues over almost three decades and resulted in the description of the Indian variant of hereditary ataxia and ultimately its genetic localization in recent times.1-20 Primary degenerative cerebellar ataxias may occur in a familial or non-familial (sporadic) form. The former, more commonly designated as hereditary ataxia (HA) may be autosomal recessive cerebellar ataxias (ARCA), autosomal dominant cerebellar ataxias (ADCA) or rarely may have X-linked recessive inheritance. The ARCAs, often designated as early onset cerebellar ataxias (EOCA) of unknown etiology, have been classified into several types by Harding.21 More recent work has helped in genetic localization in Freidreich's ataxia and some of its variants on chromosome 9 with a triplet repeat (GAA) mutation.22 But point mutation at other loci have also been identified in other forms of ARCA.23,24 The ADCAs (usually late onset), on the other hand, had been classified by Harding into 4 or more types (1,2,3 etc.).25 Advancement in genetic technology and probing facilities have helped classify the ADCAs into more than 8 types (SCA 1, 2, 3, etc.), all of which are now known to be triplet repeat disorders. These terminologies have been recommended by the Research Group on Ataxias of the World Federation of Neurology.26 The early onset sporadic forms may have AR inheritance. The late onset sporadic forms, especially with extracerebellar features, have often been grouped under the broad category of Multiple System Atrophy (MSA).27 The present report attempts to look into the pattern of primary degenerative cerebellar ataxias (familial and non-familial) amongst the ethnic Bengalee population living in the city and adjacent districts of southern West Bengal, India. MATERIAL AND METHODS In this prospective study during 1996-2000, a total of 67 cases of cerebellar ataxias and 3 asymptomatic family members with abnormal neurologic signs were examined. This figure includes fifty familial cases from 23 families (23 index cases and rest 27 affected or with abnormal neurologic signs family members) and 20 sporadic cases. Several asymptomatic cases in affected families were examined to detect abnormal neurologic signs. In particular, in all the probable ARCA cases, both parents were very carefully examined. In addition to detailed clinical examination, neuroimaging of the brain (CT or MRI) was done in all index cases and also in some of the affected family members. Electrophysiologic and evoked potential studies were done when thought necessary. In sporadic cases, especially in those without extracerebellar features, secondary causes of cerebellar degeneration (in particular, drugs, alcohol and occult neoplasia) were carefully excluded. Very detailed biochemical screening, however, could not be carried out in the EOCA cases and Vit.E levels were not measured. All such cases were given the benefit of empirical therapy with D alpha-tocopherol for at least 6 months. Molecular genetic studies were done in 42 examined patients at the Molecular Biophysics Unit, Indian Institute of Science, Bangalore or at the Crystallography and Molecular Biology Division, Saha Institute of Nuclear Physics, Kolkata. Such studies were mainly aimed at the detection of expanded trinucleotide repeats at SCA1, 2 and 3 loci and of late also included studying the SCA6, 7 and DRPLA loci. Facilities for detecting GAA repeats (in ARCA) were not available. The genetic methodologies employed had been described elsewhere.14,19,20 RESULTS Familial Cases: 50 cases from 23 families A. Early onset ataxia with probable autosomal recessive (AR) inheritance (ARCA): 18 cases Of the 12 patients (8 families), 5 patients (4 families) were Muslims with history of consanguineous marriage in the parents; the rest were Hindus with no such history. The age of onset varied from 8-18 years. The clinical features in all included ataxias, distal wasting, areflexia, extensor plantars and abnormal proprioception. No examined patient had pes cavus. None showed any oculomotor abnormality except one who had slow saccadic movement. Non-specific ST-T changes on ECG were present in all. Echocardiographic changes could be detected in 8 subjects by way of wall thickness abnormality. Electrophysiologic studies revealed abnormalities in proximal and/or distal conduction in 6 out of the 8 subjects tested. Group 2: This group included 4 patients—2 pairs of a brother-sister combination from 2 families with none else affected in the family. The parents (examined) were unaffected. The patients were negative for ADCA subtypes tested (SCA1,2,3). The age of onset in this group was between 17-24 years. The clinical features included ataxia, distal wasting, scanned speech, nystagmus (not detected in Group1), extensor plantars, and altered proprioception. The major distinctive feature was the presence of brisk tendon reflexes, especially in the lower limbs and none had any skeletal abnormality. This group merits classification separately as FA type ataxia with retained reflexes. AR inheritance seems most likely in view of the affection in one generation, with clinically unaffected parents. On neuroimaging, no patient in Group1 or 2 showed any evidence of cerebellar or brainstem atrophy. Group 3: This group included 2 patients (Muslim brothers) with clinically unaffected parents who had consanguineous marriage. No other member of the family seemed affected. AR inheritance seemed most likely and the patients tested negative for SCA 1,2,3 and 6 subtypes of ADCA. The age of onset had been very early (5-6 years) and clinical features included ataxia, optic disc pallor, bilateral lateral rectus palsy, hypo/areflexia, diminished proprioception and flexor plantars. CT showed cerebellar atrophy in one; NCV studies were normal; ECG showed non-specific ST-T changes and VEP showed increased latencies of P100 (> 110msec). Limited metabolic screen had been negative and none showed any improvement following therapy with Vitamin E for 6 months. This family was diagnosed as having early onset cerebellar ataxia (EOCA) with optic atrophy and ophthalmoplegia (non-Friedreich ARCA). B. Autosomal Dominant Cerebellar Ataxias SCA2 Families 1. SCA2 Families with extracerebellar features: Family W2: Two members of this family could be examined who showed anticipation in age of onset and severity. The index case (onset 38 years) had ataxia, dysarthria, upward gaze restriction, no nystagmus, no slow saccadic movement, hyporefexia and flexor plantars. CT showed cerebellar and brainstem atrophy. There was evidence of early motor axonopathy but Sensory Nerve Action Potential (SNAP) was normal. His affected father (onset 55 years) had only mild ataxia, no peripheral neuropathy and no oculomotor abnormality. Family W3: Only one member was examined (onset 30 years) whose father was also affected (onset 40 years). In addition to ataxia and dysarthria, he had slow saccadic movement of the eye but no gaze palsy, no evidence of peripheral neuropathy except diminished sense of vibration in legs and no pyramidal signs. CT showed cerebellar and brainstem atrophy and electrophysiology revealed normal nerve conduction. 2. SCA2 Family without extracerebellar features Both patients had prominent eyes but only cerebellar ataxia and no oculomotor, pyramidal, extrapyramidal or peripheral nervous affection. Neuroimaging revealed cerebellar and brainstem atrophy. Genetic studies of the examined patients of all the 4 families mentioned above confirmed CAG repeat expansion at the SCA2 locus. SCA3 Families 1. SCA3 families with extracerebellar features Genetic Data: Genomic DNA from the two affected members (G III 3 and G III 1) and one unaffected member were analyzed at IIS, Bangalore. In the affected individuals the lengths of repeat at SCA3 locus were 73/11 and 75/17 whereas in the unaffected member it was 26/24. Family MJ 2: Only 2 members (mother and son) in this family could be studied. The age of onset in the mother had been 30 years and that in the son 18 years. Classic features seen included ataxia, distal limb wasting, dysarthria, nasal intonation, bulging eyes, facial fasciculations, tongue atrophy, hyper-reflexia, gaze-evoked nystagmus and upward gaze weakness. Neither showed any abnormality in horizontal saccadic or pursuit movements. Electrophysiologic studies were normal. Neuroimaging revealed cerebellar and pontine atrophy. Molecular genetic study revealed CAG repeat at the SCA3 locus-repeat lengths in the mother were 83 and in the son 90. Family MJ 3: Three members in one generation could be studied. The onset age varied from 48-50 years. The index case showed a CAG repeat expansion of 69 at the SCA3 locus. Full pedigree analysis was not possible. The only extracerebellar feature was gross peripheral neuropathy-clinically and electrophysiologically (axonopathy). MR brain revealed cerebellar and parietal lobe atrophy in the index case. Family MJ 4: Only one surviving female member of this family could be studied. The onset age was 24 years and she had only truncal ataxia, gaze-evoked nystagmus and pyramidal signs in the legs. She had slightly prominent eyes but no electrophysiologic evidence of peripheral neuropathy and no ophthalmoplegia. Her mother and a maternal uncle and aunt were affected. Their onset age was mid-thirties and all three of them died between 45-48 years of age. Genetic anticipation was clearly evident. The mother's old photographs revealed progressive bulging of the eyes with advancing age and probably, she also developed distal limb muscle wasting. None of the deceased family members had any alteration in their higher mental functions (report from the father of the index case). Genetic study in the index case revealed CAG expansion at the SCA3 locus. MR scan revealed early cerebellar atrophy. 2. SCA3 Family without extracerebellar features GII 3, a female subject, presented at 64 years of age with progressive ataxia having onset at 54 years of age. The features included scanned speech, truncal and limb ataxia, nystagmus, depressed reflexes but no pyramidal signs or electrophysiologic evidence of peripheral neuropathy. No pursuit or saccadic movement abnormality was seen. GIII 1, was examined at age 42 when he had nystagmus, brisk reflexes, mild truncal ataxia but no oculomotor abnormality, peripheral neuropathy or pyramidal / extrapyramidal signs. C. ADCA of undetermined genotype Group 2: Only one family (U3) is included here separately because of some special features. The index case was 12 years old (his father was affected in his thirties and later died). Presenting features included ataxia, convergent squint, nystagmus, hyperreflexia and extensor plantars, and diminished proprioception. He had deafness and retinal pigmentory changes with posterior subcapsular cataract in both eyes. CT showed cerebellar atrophy; BAER suggested peripheral auditory pathway dysfunction; CMAP and SNAP were reduced though NCV studies were normal. Genetic screen for SCA1,2,3 and 6 were negative (clinically the features corroborated with clinical type ADCA 2 and possibly SCA7 of the genetic subtype). D. Sporadic Cerebellar Ataxias (20 cases) A. Cases with early age of onset (<20 years): This group included only 2 cases. Cases A and V had onset age at 10 years and 20 years respectively. The clinical features resembled FA type ataxia-ataxia, scanned speech, areflexia, extensor plantars and impaired proprioception but because of the absence of positive family history and the lack of facility for genetic localization, these 2 cases have been classified as sporadic cases with early onset ataxias. It may be of interest to note that case V at age 24 years, presented with cardiomyopathic features (hypertrophic non-obstructive) and the neurologic abnormalities were detected later. B. Cases with late age of onset (>30 years): This group included 18 cases (12 males and 6 females). The onset age varied from 32 years to 42 years. Progression had been slow in all patients and all except one were ambulant without support when examined even 10 years later. One case (AD) was chair-bound when first seen and died within the period of study. The clinical features in the 18 cases included: ataxia (18); dysarthria (14); pyramidal signs (6); extrapyramidal features— rigidity and resting coarse tremor (1); external ocular movement abnormality (square wave jerks - 5; slow saccades - none; abduction lag - none) and ANS disorder (one case in late stage). None showed any retinal changes or significant alteration in intellectual function during the period of study. Molecular genetic studies were conducted in 10 cases (2 at IIS and 8 at SINP). Case AD, tested at IIS, showed no expansion at SCA1,2 and 3 loci. Of the remaining 8 cases tested at SINP, case NKG (43 years) and BC (45 years) showed repeat expansion at SCA1 locus; case MD (50 years) showed repeat expansion at SCA2 locus and none of the remaining 5 cases showed any expansion at SCA1,2,3 and 6 loci. NKG (SCA1) showed electrophysiologic evidence of peripheral neuropathy, though clinically he had pyramidal signs but no extrapyramidal or ocular movement abnormality. BC (SCA1) had only cerebellar features and normal NCV studies and no pyramidal signs. M.D (SCA2) had hyper-reflexia, no slow saccadic movement, tongue fasciculation (unusual in SCA2), and no clinical or electrophysiologic evidence of peripheral neuropathy. DISCUSSION Predisposition to several genetic disorders amongst people of a particular ethnicity is well known. Hence, a clinico-genetic study of any hereditary disease must take into account the ethnic origin of the patients amongst whom the disease is being studied. In a recent communication, Wadia et al28 suggested that SCA2 is `more prevalent or more frequently recognized in India'. In a subsequent communication, Wadia et al29 stated that SCA2 was confirmed in only 6 families out of a total of 31 phenotypically similar families with ataxia and slow saccadic eye movement. Hence any conclusion about the most prevalent form of ADCA in India needs to be drawn with some degree of caution, as genotypic variation within phenotypically similar cases is well known and vice versa. The two recently published multicentric studies also need to be examined carefully. In the study reported by Saleem et al,19 out of 39 families with ADCA, genetic localization was possible only in 15. Though 10 of these were of the SCA2 type, it must not be forgotten that the families studied were of diverse ethnic origin. In the other study reported by Basu et al,20 out of 26 patients with positive family history (ADCA), SCA2 accounted for 7 patients, closely followed by SCA1 in 5 patients. This study also includes people of diverse ethnic origin. In the present study, carried out amongst ethnic Bengalees, we have failed to demonstrate a clear preponderance of any one type of SCA. Of interest may be a very recent report, also from Kolkata by Ghosh et al29 who have reported 5 patients with adult onset ataxias, presumably ethnic Bengalees, with positive family history, and found 3 cases of SCA1 type, 2 with SCA3 mutation and none with SCA2. In a multi-ethnic society, it is perhaps inappropriate to state one particular genotype to be the most common form. Globally speaking, in homogeneous population groups, certain types of ADCA have been reported to be more common than others.30-38 A. Early onset cerebellar ataxias with probable autosomal recessive inheritance (ARCA) Group 1 cases in this subgroup of ARCA with early onset, showed classic features of FA39 (12 members from 8 families). But again the term FA type ataxia has been used because of the lack of genotypic confirmation. The onset ages matched those described by Harding;39 affected members shared similar neurologic features; but none had pes cavus or any significant oculomotor abnormality (except one who showed slowing of saccadic velocity). Group 2 patients merit classification as FA type ataxia with retained reflexes (FARR)40,41 but again, genetic confirmation was lacking. The 4 patients included in the series had a slightly higher age of onset (17-24 years). Filla et al41 mentioned an earlier age of onset for FARR as compared to classical FA. However, a small number of their patients had an age of onset of about 16 years. In contrast to the series reported by Harding40 and Filla et al,41 none of our patients had any skeletal or cardiovascular abnormality. Two subjects (brothers) in Group 3 posed a diagnostic problem. AR inheritance seemed most likely because of consanguinity. The combination of optic disc pallor (associated with prolonged Visual Evoked Potential and bilateral lateral rectus palsy was unusual. Harding did not describe any specific subtype with this combination in her original classification.21 Phenotypic variants of FA are well known42 and have also been mentioned in one recent review16 (including ophthalmoplegias). Optic atrophy is, of course, a recognized feature of classical FA. Ophthalmoplegia is a recognized feature of severe Vit. E deficiency with abetalipoproteinemia. In the present case, the latter was excluded by the estimation of serum lipids and the absence of acanthocytes in the peripheral blood. At best, this group, in the absence of further detailed genetic study can be designated as an FA variant. B. Autosomal Dominant Cerebellar Ataxias (ADCA) The present series included 4 families with SCA2, 5 families with SCA3 and 3 families without any clear genotype determined. A close look at the major clinical features of the patients studied (except one case of a possible ADCA 2 family) would suggest marked inter and intra genotypic variation in the phenotype. This phenomenon has been mentioned in some of the recent reviews.16,17 The presence or absence of extracerebellar features, did not give any clue for predicting the possible genotype in the present study with ethnic Bengalee patients. Although we did not have any family with the SCA1 genotype included here, brief clinical data mentioned by Ghosh et al,29 would suggest one to the same conclusion. Only some members of one family with SCA3, showed significant extrapyramidal features. Perhaps, the presence of significant extrapyramidal features in a Bengalee patient with ataxia and a positive family history (AD type), may raise the suspicion of an SCA3 genotype. This should never be taken as a consistent or very dependable clinical marker. Similar may be the case with bulging/prominent eyes in suspected MJD/SCA3 patients. This was evident in a number of patients studied in Family MJ1 (SCA3) and in one patient reported by Ghosh et al.29 However, this was missing in other SCA3 patients studied by us and was noted in two patients in one family with SCA2 mutation (Family W4). Three other physical signs deserve special mention in studying the phenotype of ADCA patients as highlighted in recent literature. These include ocular movement abnormalities/ophthalmoplegia, peripheral neuropathy and dementia. Oculomotor abnormalities would be discussed separately. In their original report of 9 families, Wadia and Swami1 drew attention to the presence of peripheral neuropathy and distal muscular denervation as important clinical features. Subsequently, the clinical characteristics of 43 members from 29 phenotypically similar families were reported, the majority of whom had peripheral nerve dysfunction.8 Through a series of publications, Wadia and his co-workers highlighted the clinical, electrophysiologic and histological aspects of peripheral nerve dysfunction in these patients.5-7 In a recent communication, the presence of peripheral neuropathy in 14 patients from 6 families with mutation at the SCA2 locus has again been stressed.9 (sentence structure?) These 6 families were from the 31 phenotypically similar families encountered by Wadia and his co-workers till 1998. Peripheral neuropathy has also been found to be a distinctive feature in SCA2 families described from Cuba.43,44 On the basis of the above, subsequent reviews have listed peripheral neuropathy as an important phenotypic characteristic of the SCA2 genotype.16-18 In the present series, out of 6 cases of the SCA2 genotype (4 families), only 2 patients (from 1 family) showed clinical and/or electrophysiologic evidence of peripheral neuropathy. On the other hand, 8 of the 13 symptomatic patients with the SCA3 genotype showed clinical and /or electrophysiologic evidence of peripheral neuropathy and/or distal amyotrophy. In a recent communication Burk at al45 commented on the cognitive defects in German patients with the SCA2 genotype. 25% of their subjects showed clinical evidence of dementia and even in non-demented subjects, there was evidence of verbal memory and executive dysfunction. Wadia8 mentioned that dementia, if at all, had been seen late in the disease in his series. This comment, however, had been made before genotype data on some of his families was available. Similarly, Sears et al46 reported a family with olivo-ponto-cerebellar atrophy with significant dementia beginning with loss of memory and ending with a vegetative state. Again, this was before genetic data could be ascertained. In our series, none of the 6 cases of SCA2 studied (familial) showed any evidence of dementia during formal neurologic testing and on the Mini-Mental Scale. There had also been no clear history of dementia in the deceased members (affected) of the families studied. On the other hand, 2 members of a SCA3 family (MJ1) showed clear evidence of dementia even when they were still ambulant. We, therefore, fail to substantiate in our ethnic Bengalee patients, the observation of Burk et al45 that intellectual impairment represents an important part of the SCA2 phenotype. On the other hand, we noted clear evidence of dementia in some of our SCA3 genotype subjects. This aspect certainly needs further attention, especially to find any relationship between cognitive function and repeat lengths. ADCA and oculomotor disorders: Abnormalities of eye movements have been highlighted in several families with ADCA in the world literature. This aspect has been a major source of interest in studying hereditary ataxias in India. Wadia and Swami first drew attention to the presence of slow saccades in 9 Indian families with cerebellar ataxias with essentially autosomal dominant inheritance.1 In a subsequent communication, Wadia reported the occurrence of slow saccades in all 43 patients studied (from 29 families). However, the AD inheritance in all such families could not be established. Following Wadia's original publication, other reports from India also drew attention to the presence of slow saccades in families with autosomal dominant cerebellar ataxia.11,47 Wadia confirmed the slow saccadic movements with normal pursuit movement, in some of his patients by oculography.48 Later on, autopsy studies confirmed the presence of olivoponto-cerebellar atrophy in some of his patients.8 Wadia and his co-workers, subsequently demonstrated specific cell loss in the Paramedian Pontine Reticular Formation (PPRF) and attempted to explain the neural basis of the slow saccadic movements with preservation of normal pursuit.49,50 Closely similar families, with AD ataxias with slow saccades, with a common ancestry located in the Holguin Province of Cuba, have been described by Orozco et al.43,44 In our ethnic Bengalee patients in the present study, we were unable to substantiate this commonly believed notion in totality. In the present series, out of 6 familial cases of SCA2 (from genotypically proved 4 families), slow saccadic velocity was noted in only 2 patients. The only sporadic case with SCA2 genotype (vide infra) also showed no evidence of slow saccadic movement. Although oculographic studies were not performed in any case, ocular movements were very carefully looked for in all the cases studied. On the other hand, one patient with early onset ataxia of probable AR inheritance and one patient described by Ghosh et al29 (ethnic Bengalee patient) with SCA1 genotype had slow saccadic velocity. We therefore argue the reliability of the demonstration of slow saccadic movement as a clinical pointer towards SCA2 genotype, at least in ethnic Bengalee patients. In the present series, several patients with SCA3 genotype (especially in family MJI) showed gross degree of ophthalmoplegia. Ophthalmoplegia as a feature of MJD was known long before its genetic localization at the SCA3 locus was established. However, this again should not be taken as a definite clinical marker as significant ophthalmoplegia was absent in some families with SCA3 genotype in the present series. Furthermore, some cases with gross ophthalmoplegia reported by Jain and Maheshwari13 as MJD cases on clinical grounds, did not show expansion at the SCA3 locus at a later date when molecular genetic studies were carried out (Jain S. Personal communication). Possibly a hitherto unstated form of ocular pursuit movement disorder was noted in an asymptomatic girl in the family MJ1 with SCA3 genotype. Though her ocular movements during saccade were full and not slowed, during pursuit a significant `lag' was always observed in the abducting eye. This had been reported by Chakravarty earlier.14,52 On a closer look at her other family members affected with gross ophthalmoplegia, we were impressed by the finding that the restriction of movement often affected the abducting eye more rather than the adducting one. We therefore argue that, such `abduction lag' is probably an early ocular mobility disorder that members of this family with SCA3 suffered from. With the progression of the disease, more severe ophthalmoparesis supervened. A subtle degree of such `abduction lag' was also noted in one member of a family with undetermined genotype (Family U1). The `abduction lag' is probably due to a lesion at the PPRF with possible slow relaxation of the medial rectus or hypofunction of the lateral rectus muscle in the abducting eye. We feel this phenomenon needs studying in detail in patients with ataxias. In a recent communication, Rivaud-Pechoux et al53 tried to correlate eye movement abnormalities with genotype in patients with ADCA. In this study, three major criteria namely, saccade amplitude, saccade velocity and presence of gaze-evoked nystagmus permitted correct assignment of 90% of the SCA1, 90% of the SCA2 and 93% of the SCA3 patients. While we would certainly stress on the elicitation of physical signs in clinical neurological practice, we would argue against too much dependence on these for predicting genotypes in SCA patients, at least in the population that we have studied. C. Sporadic Ataxias The 2 early onset type cases had clinical features resembling FA type disease but have been included as sporadic due to lack of positive family history and our inability to characterize the genotype. Late onset cerebellar ataxias are difficult to classify. Sinha and Jha15 have preferred to continue with the older clinico-pathologic term OPCA and in 10 of their 24 cases, molecular genetic study for SCA1,2,3 and 6 had been negative. In the series reported by Futamura et al,54 in 22% of the cases (N=85) of late onset cerebellar ataxias, genetic localization could be done and SCA6 mutation was most frequently observed. In the present series, 10 out of 18 cases had genetic studies done and 2 cases of SCA1 and 1 case of SCA2 were detected. The age of onset in this group had been lower than that reported by Sinha and Jha.15 A few of our patients had pyramidal signs and one had extrapyramidal signs and later developed autonomic disturbance. However, cerebellar signs dominated in all stages of the disease. Such cases may perhaps be classified as Multiple System Atrophy with predominance of cerebellar features. It is clear that only very long-term follow-up of the cases may clarify the diagnosis in many such sporadic late onset ataxias. ACKNOWLEDGEMENTS The authors are deeply indebted to Drs. S K Bramhachari, Q Saleem (Molecular Biophysics Unit, Indian Institute of Science, Bangalore and Functional Genomics Unit, Centre for Biochemical Technology, New Delhi), and N P Bhattacharyya (Crystallography and Molecular Biology Division, Saha Institute of Nuclear Physics, Calcutta) for conducting the molecular genetic studies on the patients reported herein. REFERENCES 1. Wadia NH, Swami RK. A new form of heredo-familial spinocerebellar degeneration with slow eye movements (nine families). Brain 1971;94:359-74.
Copyright 2003 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni03070t2.jpg] [ni03070t1.jpg] |
| |||||||||

{kind=link}
{kind=link}