 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
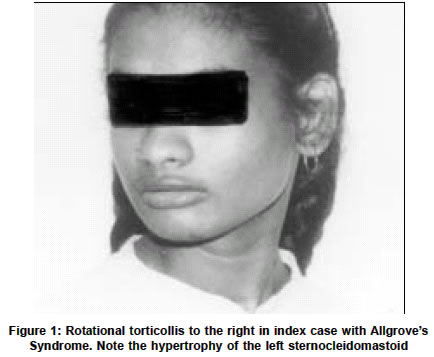
Neurology India, Vol. 51, No. 2, April-June, 2003, pp. 257-259 Case Report Two siblings with Allgrove's syndrome and extrapyramidal features A. Jacob, K. Parameswaran, A. Kishore Department of Neurology, Sree Chitra Tirunal Institute for Medical Sciences and Technology, Kerala, India. Asha Kishore
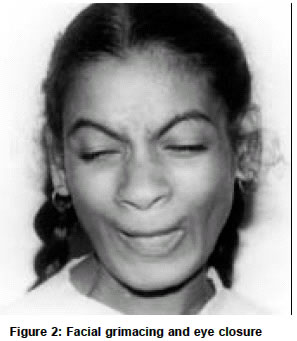
Accepted on 02.11.2001. Code Number: ni03080 ABSTRACT We report two siblings with Allgrove's syndrome and extrapyramidal features. Though various neurological abnormalities have been described in this disorder, we report the first patient of Allgrove's syndrome associated with dystonia and chorea. Key Words: Allgrove's syndrome, Alacrimia, Achalasia, Dystonia, Chorea. INTRODUCTION Allgrove's syndrome is a rare disorder clinically characterized by achalasia cardia, alacrimia and adrenocortical insufficiency (the triple A syndrome). After the initial description of two pairs of siblings in separate families by Allgrove et al, approximately 70 cases have been described.1 Autonomic dysfunction was subsequently found to be a frequent association, hence the name "4 A syndrome" has been proposed.2 The presence of palmoplantar hyperkeratosis, neurological manifestations, short stature, microcephly and osteoporosis indicate the multisytem nature of this rare disorder.3-9 Extrapyramidal involvement is rare and parkinsonism is the only manifestation reported so far.9 We report a unique familial Allgrove's syndrome in two siblings who exhibited dystonia and chorea. CASE REPORTS An 18-year-old lady was referred for evaluation of involuntary movements of 5 years duration. She was the eldest child of a first-degree consanguineous marriage and had a normal perinatal history. She had delayed motor, mental and language milestones, which gradually improved over the years. She could speak fairly well and was almost independent in all activities of daily living by the age of 10 years. She had dysphagia with regurgitation of food from early childhood that was diagnosed as achalasia cardia at the age of 12 years. Balloon dilatation relieved her dysphagia. According to her mother she had never cried in her life and her mouth was always dry. She gave no history of postural syncope, palpitation, disturbance of sweating, diarrhea or constipation. At 13 years of age, sustained neck turning to the right side was observed. Attempts to turn her neck to the left were painful. In addition, she had random sudden jerks of the neck to the right side. There was no posturing of any other body parts, tremulousness, slowness or stiffness of the limbs. These involuntary movements were absent during sleep. There was no history of reduced vision, hearing, seizures, stiffness, weakness, wasting, unsteadiness or numbness. The examination of the index case revealed normal heart rate and blood pressure. There was no postural hypotension. She had icthyosis of both lower limbs but no skin hyperpigmentation. Her conjunctiva and oral cavity were dry. There was no KF ring or cataract. Fundi were normal. Formal IQ testing showed score of 48 by Wechsler Adult intelligence score. Pupils were irregular, asymmetric in size (right = 3mm, left =-5mm) with no reaction to light or accommodation. Extraocular movements were normal. Her jaw jerk was brisk. There was no jaw, facial or palatal weakness. Her left sternomastoid was hypertrophic and she had rotational torticollis to the right (Figure 1). Her right shoulder was elevated and right trapezius was hypertrophic. In addition she also had frequent, brief, jerky head movements to the right occurring at rest and increasing on attempts to turn the neck to the left. These movements worsened with attention and excitement. There was also frequent facial grimacing associated with closure of eyes (Figure 2). Voluntary suppression of these movements couldn't be checked because of poor cooperation and her low IQ. She also had intermittent, random predominantly distal, jerky movements of the upper and lower limbs, which appeared semi-purposive and migratory in nature. She had bilateral striatal toes. Her muscle bulk and power were normal. All deep tendon reflexes were brisk along with bilateral finger flexion and positive Hoffmann's sign. Cerebellar and sensory systems were normal. The index case's younger sister had similar neurological findings though milder in intensity. She had mental subnormality, achalasia (operated), alacrimia and reduced salivation along with neck dystonia and facial grimacing. Hemogram and routine biochemical tests, and chest X-ray were normal. Serology for syphilis, rheumatoid factor, SLE and antiphospholipid antibodies were negative. Acanthocytes were not seen in repeated examinations of peripheral smear. Slit lamp examination for KF ring was negative and serum ceruloplasmin was normal. ENMG was normal. MRI brain didn't reveal any abnormality. Serum cortisol at 8 am was 6.4 micro gram/dl (Normal 5 to 29 micro gram/dl). Schirmer's test was negative suggesting reduced lacrimation in both eyes. Basal values were 0 mm in both eyes at 5 minutes and reflex secretion was 0 mm in the right eye, and 2 mm in the left eye at 2 minutes (normal is more than 5 mm). Tests for salivatory function showed markedly reduced salivation (1.38ml/5mts, control-19.9 ml/5 minutes). Heart variability with respiration was normal. Max R-R/Min R-R was 1.35 (Normal>1.2). Normal heart rate variability was present—79-98 bpm (Normal> 15bpm). Heart rate response to standing was normal. 30:15 ratio was 1.1 (normal>1.04). DISCUSSION Our patient and her sibling had the classical features of Allgrove's syndrome—achalasia, alacrimia along with autonomic dysfunction. Their neurological manifestations included the already well-known features such as mental retardation and corticospinal dysfunction. In addition, they had prominent neck dystonia, facial and limb chorea, which have hitherto not been reported in this syndrome. The only extrapyramidal feature of Allgrove's syndrome described so far in the literature is parkinsonism in brothers who had in addition, dementia, and pyramidal signs.5 The levels of CSF homovanillic acid and 5-hydroxy indole acetic acid (the major metabolites of dopamine and serotonin in the brain) were low in them. Positron emission photometry scans in the older boy showed reduced uptake of 18-F-Flurodopa in the striatum. However, treatment with levodopa up to 800 mg per day produced only mild improvement in their parkinsonism. The lack of clinical response to levodopa could suggest additional involvement at the level of striatal dopamine receptors or pathways downstream in the basal ganglia circuits. The presence of dystonia and chorea in our patients indicate basal ganglia involvement. We did not have facilities for PET studies to examine the pre- and postsynaptic dopaminergic systems in our patient. The abnormality reported in neuroimaging in this condition is periventricular brain heterotopias. It has been suggested that heterotopias could indicate abnormality of gene (genes) causing heterotopias or neurotrophic activity during pre- and postnatal life and that multiple or contiguous genes may be involved in Allgrove's syndrome. This syndrome has been mapped to chromosome 12q 13 near the type II keratin gene cluster.3 Intra and interfamilial variability of the severity observed in Allgrove's syndrome implies a highly variable expression of the defective gene. In conclusion, the neurological manifestations of dystonia and chorea in the two siblings we have reported here are believed to be unique. The neurochemical and neuropathological basis of basal ganglia dysfuntion in this disorder are unknown. It may be worthwhile exploring the multiple/ contiguous gene theory of this disease by including the dopamine receptor genes since dystonia, chorea and levodopa-unresponsive parkinsonism could all result from abnormalities in the striatal dopamine receptors of the basal ganglia circuits. REFERENCES 1. Allgrove J, Clayden GS, Granto DB, Macaulay JC. Familial glucocortiod deficiency with achalasia of the cardia and deficient tear production. Lancet 1978;1:1284-6.
Copyright 2003 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni03080f2.jpg] [ni03080f1.jpg] |
| |||||||||

{kind=link}
{kind=link}