 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
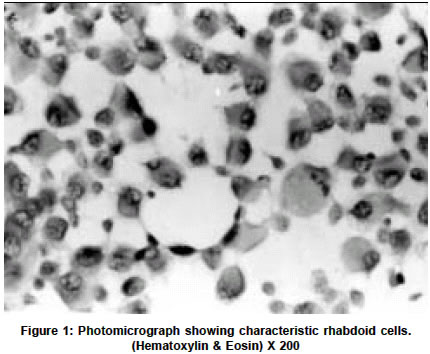
Neurology India, Vol. 51, No. 2, April-June, 2003, pp. 273 Short Report Rhabdoid tumor of the thalamus R. Kachhara, T. M. Retnam, S. Kumar, S. Nair, R. N. Bhattacharya, T. Krishnamoorthy,* V. V. Radhakrishnan** Departments of Neurosurgery, *Radiology and **Pathology, Sree Chitra Tirunal Institute for Medical Sciences and Technology Trivandrum, India. V. V. Radhakrishnan Accepted on 11.04.2002. Code Number: ni03086 ABSTRACT Rhabdoid tumors of the central nervous system are uncommon tumors. About 188 cases have been reported in the literature so far. In this report, we describe a case of a rhabdoid tumor of the thalamus in a 35-year-old male patient. Light microscopic and immunohistochemical features are discussed and the relevant literature reviewed. Key Words: Rhabdoid tumor, vimentin, immunohistochemistry. Rhabdoid tumour of the central nervous system (CNS) is an uncommon tumor. A survey of the literature till 2000 revealed 188 published cases of rhabdoid tumors of the CNS.1 We describe clinical, light microscopic and immunohistochemical features of a rhabdoid tumor of the thalamus. CASE REPORT A 35-year-old male patient was admitted with a history of bifrontal headaches and numbness of the left half of the body for 2 months. Numbness started in the left great toe and it gradually ascended within 2 weeks to involve the entire left half of the body. A month prior to admission he developed weakness of left-sided limbs. On examination, he was fully conscious and oriented. He had left spastic hemiparesis and left hemianesthesia. The rest of the examination was normal. Magnetic resonance imaging (MRI) of the brain revealed a well-circumscribed mass lesion in the right thalamic region which was hypointense on T1-weighted images and hyperintense on T2-weighted images. The periphery of the tumor enhanced on gadolinium administration. There was minimal perilesional edema. Through a craniotomy, the necrotic part of the tumor was radically removed. The periphery of the tumor was firm and merged with the adjoining normal brain. The patient had an uneventful postoperative course. While on radiotherapy, the tumor recurred and involved the right lateral ventricle. Hematoxylin and eosin-stained sections showed a diffuse homogenous neoplasm made up of large round or oval cells with eccentric hyperchromatic or normochromatic nuclei. The normochromatic nuclei of the neoplastic cells contained prominent nucleoli and abundant eosinophilic cytoplasm. Mitotic figures (1-2 per high power field) were also seen (Figure 1). Some of the large neoplastic cells showed tapering cytoplasmic processes resembling "tadpole". These large neoplastic cells were regarded as rhabdoid cells. At the periphery of the rhabdoid cells, there were loosely textured spindle cells and they were arranged in compact fascicles. Multiple discrete foci of necroses were seen within the tumor. In the entire section neither neuroectodermal nor epithelial components were identified. Immunohistochemistry for glial fibrillary acidic protein (GFAP) and vimentin were performed in paraffin section of the tumor. The majority of the rhabdoid cells (> 80 %) showed positive immunoreactivity for vimentin. These rhabdoid cells had negative immunoreactivity for GFAP. Based on light microscopic and immunohistochemical studies, a diagnosis of rhabdoid tumor of the thalamic region was made. DISCUSSION This case showed characteristic light microscopic and immunohistochemical features of a rhabdoid tumor. Positive vimentin immunoreactivity strongly supported the diagnosis of rhabdoid tumor. The light microscopic features of rhabdoid tumor may mimic gemistocytic astrocytoma or glioblastoma and vimentin immunohistochemistry can distinguish rhabdoid tumors from these tumors. In our case, apart from the rhabdoid cells, there were also mesenchymal components in the form of spindle cells. The precise histogenesis of the rhabdoid cells is not known. However, histiocytic2,3 myogenous4,5 mesenchymal,6 and meningeal,7 origins have been postulated. Rorke et al8 suggested these tumors as atypical teratoid / rhabdoid because there was an admixture of primitive neuroectodermal, mesenchymal and epithelial elements in their cases. However, histological components in the rhabdoid tumors appeared different from more familiar teratomas and furthermore, the neoplastic cells in the rhabdoid tumors gave consistent negative staining for germ cell markers. The true incidence of rhabdoid tumors of the CNS is difficult to obtain as these tumors have generally been misdiagnosed. A review of the literature revealed that 94 % of rhabdoid tumors occurred in the pediatric age group and 6% in adults. 52 % of rhabdoid tumors of the CNS have been reported to occur in the infratentorial region and 39% were reported in the supratentorial region. Pineal, spinal and multifocal cases accounted for 5%, 2% and 2% respectively.1 There are neither clinical nor imaging features specific for the diagnosis of rhabdoid tumors and the diagnosis is often based on histopathology. Rhabdoid tumors of the CNS are regarded as aggressive malignant tumors. Majority of the cases succumb within a year of diagnosis.8 The maximum period of survival has been reported as 4-6 years.1 Postoperative radiotherapy is advocated even if total resection is done. ACKNOWLEDGEMENTS The authors wish to thank Prof K Mohandas, Director, Sree Chitra Tirunal Institute for Medical Sciences & Technology, Trivandrum for the kind permission to publish this case. REFERENCES 1. Kleihues P, Cavenee WK. Atypical teratoid / rhabdoid tumours. In: Pathology and Genetics of tumours of Nervous systems. Lyon: IARC Press; 2000. Chapter 8. pp. 145-8.
Copyright 2003 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni03086f1.jpg] |
| |||||||||

{kind=link}