 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
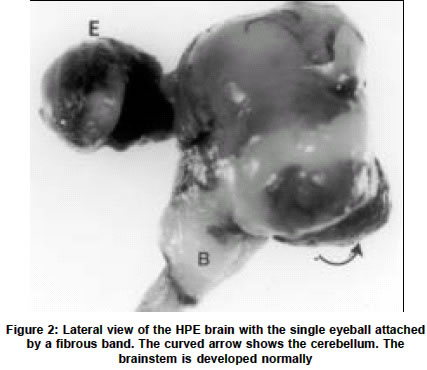
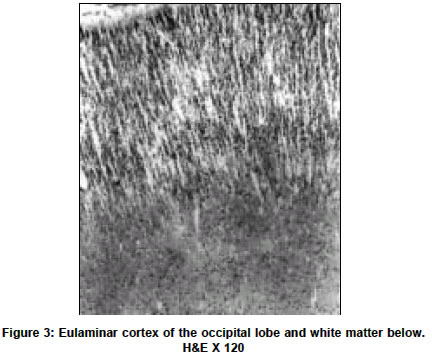
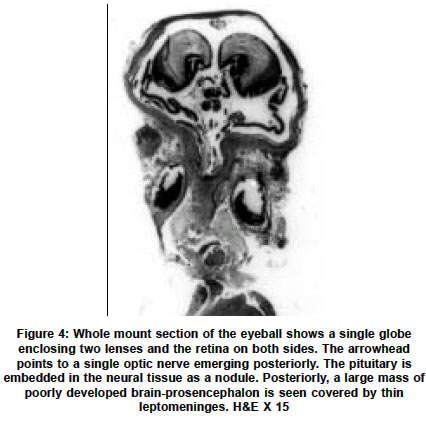
Neurology India, Vol. 51, No. 2, April-June, 2003, pp. 279-282 Short Report Holoprosencephaly with cyclopia - Report of a pathological study N. Arathi, A. Mahadevan, V. Santosh, T. C. Yasha, S. K. Shankar Department of Neuropathology, National Institute of Mental Health and Neurosciences, Bangalore-560029. S. K. Shankar Accepted on 12.01.2002. Code Number: ni03090 ABSTRACT A rare case of a lobar holoprosencephaly with cyclopia, associated with non-nervous system anomalies is being reported. Key Words: Holoprosencephaly, Arhinencephaly, Clopia cyclopia, Craniofacial dysplasia. Holoprosencephaly (HPE) with cyclopia is an infrequent congenital anomaly of the forebrain system. We describe a case of cyclopia with HPE, with associated non-nervous system anomalies, detected in an aborted fetus. CASE REPORT An aborted female fetus of 16 weeks gestation was examined. This was the second pregnancy for the 26-year-old mother, the first pregnancy having produced a normal, healthy child after a full term. There was no history suggestive of a teratogenic insult. Prenatal ultrasound scanning revealed multiple anomalies, considered nonviable for the fetus and hence the pregnancy was terminated by induction. The aborted fetus was fixed in 10% formalin and dissected. GROSS EXAMINATION The crown rump length of the fetus was 16 cm (normal for the gestational age). The most striking anomaly was a single eye in the mid-forehead (cyclopia) (Figure 1). In the face, there was no nasal aperture nor proboscis in the midline. The external ears were normal. No cleft lip or cleft palate was noted, but there was micrognathia and a prominent tongue. The anterior abdominal wall was deficient causing the internal organs, enveloped in a thin transparent membrane, to herniate out. The upper limbs had short forearms, but long tapering fingers. The lower limbs were grossly retroflexed at the hip. The thoracic cavity was communicating with the abdominal cavity due to posterior diaphragmatic eventration. The lungs were hypoplastic and thymus was not found. The heart was grossly malrotated, the apex pointing upwards. Separate atria could not be made out clearly. The inferior vena cava from the liver was seen terminating into the atrium on the left. A single vessel was seen arising from one of the lungs, entering into the atrium, while the main pulmonary artery could not be identified. From the ventricular chamber, the aorta curved to the right to reach behind the superior vena cava. Further details could not be appreciated. In the abdominal cavity a severe degree of kyphoscoliosis was noted, the abdominal organs adherent and malrotated, forming a mass. The stomach and the esophagus were normal. The duodenum, proximal to the atretic segment of the jejunum, was dilated. The vermiform appendix could not be identified. The large intestine was normal. The liver, spleen and pancreas were normal. The kidneys appeared cystic, dilated and dysplastic and the rest of the genitourinary system could not be traced. The adrenal glands were small and located at hilum of liver. The vault of the microcephalic skull revealed fusion of the frontal bones, obliterating the anterior fontanellae. The malar bones appeared to have fused to form the floor of the orbit, the fused frontal bones, without frontal bossing, forming the orbital roof. A single eyeball was seen, covered by eyelids and a single cornea. The base of the skull was shallow, especially the middle cranial fossa. In the midline cristagalli and cribriform plate were absent. The foramina of the middle cranial fossa and the sella were not clearly discernable. Posteriorly, the clivus was prominent and straight. The petrous temporal bone on both sides was prominent. The brain, covered by thin leptomeninges, was small, filling the skull, and weighed 80 gm. The falx cerebri was absent, while the tentorium cerebelli was seen, the insertion being elevated due to bony anomalies. The eyeball located in a single orbit was attached to the forebrain by a fibrous band. Two separate cerebral hemispheres were not discerned. The single cerebral hemisphere was smooth, with no lobes or interhemispheric fissure _ features that are characteristic of "alobar holoprosencephaly" (Figure 2). A hypoplastic cerebellum was seen straddling over the posterior part of the cerebral hemisphere, covering the occipital poles and displaced more to the right side. The brainstem and spinal cord were normal till the cervical segments. The caudal end could not be traced due to the vertebral anomalies. At the base of the brain, the olfactory bulbs, tracts, and the optic chiasma were absent, while other cranial nerves were discernable as hairy thin strands. On slicing the brain the lateral ventricles were formed but distorted and compressed. The corpus striatum and thalamus were malformed, but found on both sides of the anterior third ventricle that was seen as a slit. The aqueduct could not be discerned, while the fourth ventricle was patent, with normal pons, medulla oblongata and cervical cord. On slicing, the two cerebellar hemispheres bridged by vermis and the white matter could be seen. On histological examination, the leptomeninges appeared to be less vascular for the gestational age. The cortex corresponding to the frontoparietal zone revealed granular cortex with irregular lamination and focal hypocellular areas, suggesting migration disorders. Posteriorly, the cortex was eulaminate showing large Betz cells of the parietal cortex and granular koniocortex of the occipital lobe at the pole (Figure 3). The cortex bordering the ventricular lining inferiorly showed architecture reminiscent of entorrhinal area. The corpus striatum had dense masses of immature neurons, while the diencephalon had relatively mature neurons. The brainstem and cerebellum revealed normal cytoarchitectural maturation. In the white matter, myelination gliosis was evident. GFAP immunostaining labeled the subependymal glia, but not the radial glia. Immunostaining with antibody to phosphorylated H and M neurofilament peptides, highlighted only a few of the long fiber tracts traversing the brainstem and cerebral cortex, indicating the immature nature of the developing axons. In the subarachnoid space behind the fused dorsal thalamic masses, an immature pineal gland was located. No stigmata like microglial nodules, focal calcification of parenchyma or vessels were found anywhere in the brain or other organs, excluding possible infection by TORCH complex. The choroid plexus was histologically normal. Histological examination of the cyclopia revealed the eyeballs to be fused into one scleral globe of normal size, with a single optic nerve emerging from the posterior part. Anteriorly, a single cornea was seen. Two hyalinized acellular lenses were seen, the ciliary body attached along the lateral margin of both lenses, with a single pupillary aperture. The two lenses were separated by a partial central mesenchymal band arising from the vascular vitreous (Figure 4). The retina was immature and had redundant folds and islands of rests with rosettes. In the dura behind the globe, extraocular retinal islands were seen, enclosed in a vascular stroma. The normal laminar nature of the retina was noted within the globe, while the extraocular retinal rests were immature. Embedded in the arachnoid, rudimentary adeno and neurohypophysis were seen, posterior to the optic nerve. The optic nerve continuity to the chiasma was not discernable, while the optic tract close to the hippocampal fissure was seen. DISCUSSION Cyclopia is the most grotesque and repelling eye deformity among the median facio-cerebral developmental deformities. Ulysses and his men were tormented by the one-eyed giant Polyphemus in the land of Cyclops (Odyssey by Homer). While these median facial anomalies can be diagnosed at a glance, they predict a severe degree of cerebral anomaly like HPE. During embryogenesis, the prechordal mesoderm not only forms the median facial bones but also induces the rostral neuroectodermal differentiation and morphogenesis. Defects in the prechordal mesoderm due to mechanical, genetic or environmental teratogens can lead to the arrest or malformation of the facial bones and organogenetic cleavage of the prosencephalon. The mandible may also be affected along with other facial bones, leading to micrognathia. In extreme cases, when the prosencephalon fails to cleave sagittally into cerebral hemispheres or transversely into telencephalon and diencephalon or horizontally into the optic and olfactory bulbs, the neural tube vesicles remain holistic as alobar HPE with grave prognosis and high mortality. In the intermediate and milder end of the spectrum, one may find semilobar or lobar prosencephaly, associated with correctable facial anomalies. DeMyer et al2 suggested that the type of brain malformation can be predicted by the facial development; the closer it approaches to the normal, so does the brain. However, several other studies have implied that this is not necessarily so. Miller3 reported a case with severe degree of facial anomaly, while the brain revealed absence of olfactory bulbs as the only abnormality, contrary to the expected HPE. This suggests that other modulatory factors are involved in the stages of morphogenesis of embryonally related structures. Hydrocephalus could be present with HPE, which is of importance in neurosurgical practice. Jellinger et al4 reported associated hydrocephalus in 52% HPE cases. Interestingly, in a review of 100 cases of HPE it was noted that hydrocephalus was not prevalent in patients with apparent facial dysmorphias but common in cases without facial stigmata (Osaka et al1). Similarly, hydrocephalus is uncommon in alobar HPE, as in the present case, but common in the semilobar and lobar types. HPE cases are hopelessly demented and succumb in infancy or early childhood. It may be associated with cortical dysplasia like agyria, micropolygyria and grey heterotopias, and displaced hippocampus indicating related neuronal migration disorders. The eye changes in HPE may vary from gross to subtle. In the 22 arhinencephalic infants studied by Karseras et al,5 no case of cyclopia was found and they opined that eye-brain anomalies may not be necessarily parallel indicating dissociation in the temporal evolution of these structures. Systemic examination of the HPE cases may reveal varying degrees and patterns of extracerebral abnormalities, as noted in the present case. Jellinger et al4 found associated anomalies in other organs in 53.5% cases, the commonest being in the gastrointestinal system. The authors stated that there is considerable heterogeneity of associated malformations in the CNS and even greater outside the CNS, due to widespread but varied developmental disturbances. The etiology of HPE midline defects in man is not known. Association of the disorder with various recessively inherited syndromes suggests a genetic basis. In general, HPE with few or no extracerebral systemic anomalies have normal karyotype. Those who have extracephalic anomalies along with HPE are usually found to have trisomy 13 or 18 and triploidy.6-11 However, exceptions to these observations exist. In the present case, no karyotype studies were carried out on the fetus, as it was received in formalin, several days later. To account for the evolution of cases with normal karyotype, maternal rubella, toxoplasmosis, alcoholism, diabetes, and drug treatment have been implicated as the etiological agents. Benwara Raghbir et al10 suggested a role of maternal ingestion of salicylates in the evolution of cyclopia and other anomalies. Hereditary pattern and the experimental model for cerebral malformations suggest that these anomalies are polyetiologic defects, the manifestation correlating with the temporal stage of morphogenesis and the interaction with the etiologic agent. In view of the genetic basis and polypharmacy in general practice, it is essential to offer timely counseling to the parents. In addition, the study provides insight into the events modulating the morphogenesis of the fetus. ACKNOWLEDGEMENTS The authors wish to acknowledge Dr. Anita Agarwal, Consultant Obstetrician & Gynecologist, Amar Jyothi Nursing Home, Indira Nagar, Bangalore for providing the fetus for study following informed consent. REFERENCES 1. Osaka K, Matsumoto S. Holoprosencephaly in neurosurgical practice. J Neurosurg 1978;48:787-803.
Copyright 2003 - Neurology India. Also available online at http://www.neurologyindia.com The following images related to this document are available:Photo images[ni03090f2.jpg] [ni03090f1.jpg] [ni03090f4.jpg] [ni03090f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}