 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
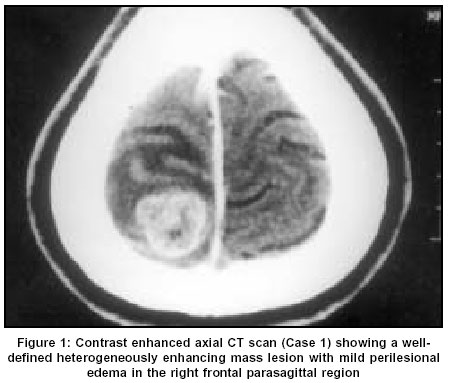
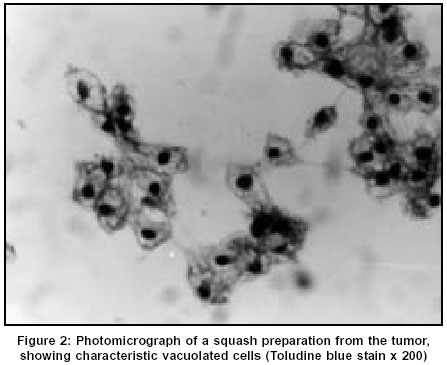
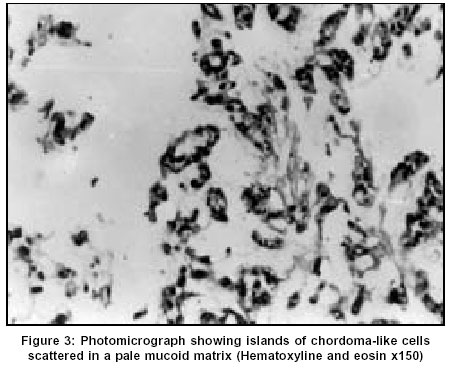
Neurology India, Vol. 51, No. 4, October-December, 2003, pp. 522-524 Case Report Chordoid meningioma: A report of two cases Varma DR, Rao BR, Parameswaran S, Gupta AK, Joseph S, Radhakrishnan VV Departments of Pathology, Sree Chitra Tirunal Institute for Medical Sciences and Technology, Thiruvananthapuram Code Number: ni03166 Abstract Chordoid meningioma is an uncommon histopathological variant of meningioma. We report 2 cases of chordoid meningioma occurring in adult patients.Introduction Meningiomas are some of the most common intracranial tumors. The 1993 World Health Organization classification of CNS tumours described 14 distinct histopathological variants; chordoid meningioma is one of them. A literature survey till 2001 revealed that apart from 2 large series,[1],[2] the majority of the documented cases are individual case reports.[3],[4],[5],[6] In this report, we describe 2 additional cases of chordoid meningioma, highlighting the clinical, neuroradiological and light microscopic features.Case Reports Case 1 The patient underwent a right frontal craniotomy for resection of the tumor. The lesion was seen as a firm globular mass which was adherent to the dura just lateral to the superior sagittal sinus. There was a well-defined gliotic plane around the tumor and the tumor could be easily separated from the brain surface. The tumor was excised in toto and sent for histopathological examination. The patient was asymptomatic during the one-year follow-up after surgery. CT study performed 6 months after surgery showed complete resolution of the edema without any residual mass lesion. Case 2 CT of brain revealed a well-defined heterogenous hypodense mass lesion in the right frontal parasagittal region. The lesion had a broad base towards the anterior third of the falx cerebri. Heterogenous enhancement was seen on post-contrast study. Moderate perilesional edema was noted, causing mass effect with subfalcine herniation. The patient underwent a right pericoronal parasagittal craniotomy resection of the tumor. The mass seen in the parasagittal region along the anterior third of the falx was soft to firm in consistency. There was a well-defined plane of cleavage around the mass. Total excision of the lesion could be performed. Histopathological Findings The entire specimens obtained at surgery from both patients were subjected to light microscopic and immunohistochemical studies. In both cases, sections revealed sheets and lobules of epithelial-like cells which were seen scattered in a pale basophilic myxoid matrix [Figure - 3]. Some of these cells exhibited characteristic cytoplasmic vacuolization. Imperceptibly merging with these epithelial-like cells, were concentric whorl and lobule-like arrangements of cells which were characteristic of meningioma. In both cases, foci of lymphoplasmacytic infiltration were also seen. The specimen from the first patient also showed atypical nuclear changes with nucleolar prominence, occasional mitotic figures and focal nodular neural invasion. In both cases, the epithelial-like cells scattered in the basophilic myxoid matrix were immunoreactive for vimentin and epithelial membrane antigen. These cells showed negative immunostaining for glial fibrillary acidic protein. Based on the light microscopic and immunohistochemical staining characteristics, a histopathological diagnosis of chordoid variant of meningioma was made in both cases. Discussion Kepes et al[1] for the first time, described a distinct meningeal tumor occurring in children and this was associated with systemic manifestations such as refractory microcytic anemia, hypergammaglobulinemia and angiofollicular lymphoid hyperplasia (Castleman Disease). Histopathologically, these tumors were composed of spindle or epithelial cells forming chordoma-like clusters and were scattered in a myxoid matrix. Prominent lymphoplasmacellular infiltration was also a feature in these tumors. Kepes et al also documented in their study, that administration of corticosteroids was usually associated with complete resolution of systemic manifestations and a significant decrease in the tumor volume. In a large series of 42 patients, Couce et al[2] observed that none of their cases had any systemic manifestations as reported by Kepes et al. The mean age of patients in the series of Couce et al was 44 years and hence they expressed the possibility that the systemic manifestations associated with this tumor are limited to chordoid meningiomas occurring in the childhood. The 2 patients described in this report were young adults and their main symptom was headache. The first patient also had episodes of generalized tonic-clonic seizures. There were no features suggestive of Castleman disease in either of our patients. Neuroimaging features in both our patients were more or less similar to that of typical meningiomas. However, in the first patient, the parasagittal location of the tumor gave a false appearance of an intra-axial lesion in the axial CT scan. In light of the elevated erythrocyte sedimentation rate, this was interpreted as a tuberculoma. Progression of neurological illness in this patient warranted a repeat CT study which revealed the features of a meningioma. Following a complete surgical excision, the patient became asymptomatic. CT scan performed at 6 months after surgery showed absence of edema and ruled out any residual lesion. The patient was on follow-up for 1 year and has not reported any episodes of headache or seizures. Both patients have been advised to present themselves for long-term follow-up. The precise diagnosis of chordoid meningioma can only be made with the help of an accurate histopathological analysis. Though the histopathological features are distinctive, these tumors should be distinguished from chordoma, chondrosarcoma and metastatic mucinous carcinoma. The identification of the classical meningioma component and the demonstration of membrane immunostaining in these tumors will establish the diagnosis of chordoid meningioma. However, difficulty in making the diagnosis can still be encountered, particularly for those tumors which arise from the base of the skull. In these cases, differentiation from chordoma may be extremely difficult. The recurrence rate in chordoid meningioma is reported to be very high. Couce et al[2] observed recurrence in all those tumors that were subtotally resected. A possible explanation for the high rate of recurrence could be related to the mucoid quality of its stroma which mechanically facilitates the spread of the neoplastic cells. This explanation is supported by the fact that all the recurrent tumors in their study were associated with a predominance of a mucin-rich chordoid pattern. Periodic follow-up and imaging studies after surgery are indicated if resection has been subtotal or if tumor recurrence is suspected on clinical grounds. References
Copyright 2003 - Neurology India Free full text also available from: http://www.neurologyindia.com/article.asp?issn=0028-3886;year=2003;volume=51;issue=4;spage=522;epage=524;aulast=Varma The following images related to this document are available:Photo images[ni03166f2.jpg] [ni03166f1.jpg] [ni03166f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}