 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
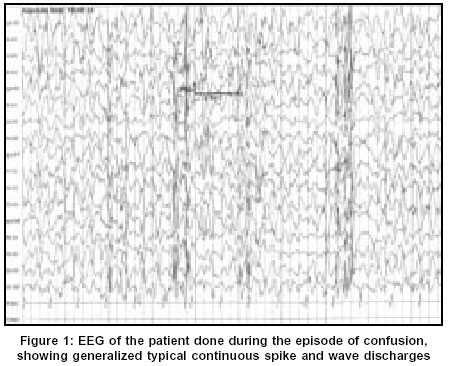
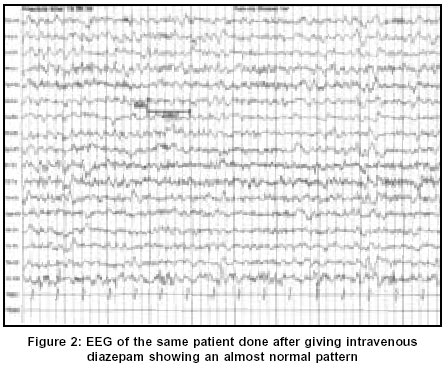
Neurology India, Vol. 51, No. 4, October-December, 2003, pp. 531-533 Case Report Non-convulsive status epilepticus: A rare presentation of juvenile myoclonic epilepsy Chemmanam T, Pandian JD, Singh YP, Pandhi M Department of Neurology, Genetics, Christian Medical College and Hospital, Ludhiana - 141008 Code Number: ni03169 Abstract We present a case of a boy with juvenile myoclonic epilepsy (JME) who presented with features of non-convulsive status epilepticus (NCSE). This case highlights the fact that NCSE, even though not a common occurrence in JME, should be kept in mind when a patient with previous history of seizures presents with subtle changes in sensorium with no obvious cause.Introduction Juvenile myoclonic epilepsy (JME) is one of the most common age-related idiopathic generalized epileptic syndromes. JME may manifest with different seizure types, the most common being myoclonic jerks. The other most commonly noticed seizure types are generalized tonic clonic seizures (GTCS), and absence seizures.[1],[2] Furthermore, there have been case reports of JME presenting with non-convulsive status epilepticus (NCSE).[3],[4],[5] We present the case of a boy with JME who presented to us with NCSE.Case Report This 16-year-old boy presented with alteration in behavior and inability to speak for one day. This boy was in good health till 8 months back when he started developing sudden jerky movements of both upper limbs. These jerks were noticed to be more on awakening in the morning and he used to drop things held in his hand. Four months later, he had one episode of GTCS, which lasted for a few minutes. This was followed by a post-ictal confusional state, which lasted for about 10 minutes. He had 2 further episodes of GTCS, 1 month and 2 months after the initial episode. He had history of reduced sleep prior to the first 2 episodes of GTCS. The exact details of the anticonvulsant drugs that he was on were not available. One day prior to admission to this hospital, he was found to be lethargic, less responsive and disoriented. Over the next few hours he was found to be more drowsy and confused and unable to speak. He did not have any GTCS at that time, even though the boy's parents noted occasional myoclonic jerks. There was no history of fever, headache or stiff neck. He did not have any history of seizure in early childhood or in the neonatal period. His developmental milestones and school performance was normal. There was no previous history of meningitis, encephalitis, trauma or other systemic symptoms. There was no history of exposure to drugs or toxins His younger brother experienced GTCS at the age of 13 years, which was followed 3 months later by myoclonic jerks on awakening. His younger sister also had onset of GTCS and myoclonic jerks on awakening at 10 years of age. Seizures are well controlled with sodium valproate in both the siblings. There was no history of consanguinity in the parents. Examination revealed a young boy who was drowsy and disoriented. He was not able to follow simple commands. He had a vacant stare but was moving all four limbs spontaneously. Occasional myoclonic jerks were noted in both upper limbs and lower limbs. Cranial nerve examination was normal. Deep tendon reflexes were bilaterally brisk and both plantar responses were extensor. He was afebrile and there were no meningeal signs. There were no other abnormalities on neurological as well as systemic examination. Optic fundi were normal on examination. Investigations revealed a normal hemogram. Metabolic parameters including serum creatinine and blood urea, serum electrolytes, serum calcium, magnesium and phosphorous were normal. CT scan of brain with contrast was normal. Other investigations like serum lactate, HIV and thyroid function tests were normal. Digital Video EEG was done during the episode of confusion using Nicolet Alliance NT machine, which showed generalized 2-3 Hz continuous spike and wave discharges [Figure - 1]. The EEG changes dramatically improved after the initial 2 mg of intravenous diazepam [Figure - 2]. He started having significant improvement in his sensorium and became more responsive after the next 2 mg dose of diazepam. He fulfilled the clinical and EEG criteria for NCSE.[6]-[7] He was initiated on midazolam infusion and he started talking and responding to commands within a few hours. The Mini mental status examination (MMSE) was normal (30/30). He was discharged on 1 g per day of sodium valproate. Follow-up MMSE and neurological examination was normal at 3 months. A repeat EEG showed generalized 3-4 Hz spike and wave discharges with normal background activity and photoparoxysmal response. EEG of both the siblings showed generalized 3-4 Hz spike and wave discharges. Discussion JME is an idiopathic age-related generalized epileptic syndrome, which is characterized by myoclonic jerks, absences and GTCS on awakening.[1],[2] This boy who presented with recent onset of myoclonic jerks and GTCS without any cognitive decline and with the EEG findings, fits into the diagnosis of JME.[8] There is a positive family history in this case and this is in accordance with several studies, which show a positive family history in 17 to 50 % of patients, with the risk of occurrence being in direct proportion to the degree of relation.[2] Other potential causes of myoclonic epilepsy like progressive myoclonic epilepsy (PME) could be ruled out by the absence of progressive cognitive decline, complete recovery with treatment and absence of evidence of the involvement of other parts of the nervous system.[9] The occurrence of NCSE in JME is not very common. There have been few case reports in the literature on the occurrence of NCSE in JME.[3],[4],[5] This has been reported in all age groups ranging from 10 years to 69 years of both sexes. Dziewas et al reported 4 patients with JME presenting with NCSE and predicted a prevalence of 5.8% and an incidence of 1.2% per year. All the 4 patients had history of myoclonic jerks, GTCS and absence seizure.[3] Two patients of JME presenting with NCSE have been reported from Japan. They had mild lethargy, slowing of responses and trembling of eyelids, but EEG revealed continuous generalized polyspike-waves.[4] The association between NCSE and JME is not very common the other way also. Among the various causes of NCSE, JME is not a very common cause. Tomson et al in a retrospective study of 32 patients with NCSE could identify only 1 patient with JME and 6 patients with absence seizures.[10] The NCSE in JME responds promptly to treatment with benzodiazepines unlike in cases of complex partial status.[11] The patient in the present case showed dramatic improvement with diazepam. Inadequate and inappropriate medication has been reported to worsen the seizures in JME.[11] This may also be the case in our patient, but this could not be substantiated since the identity of the antiepileptic drug that he was taking was not available. However, a detailed evaluation in this case did not reveal any other cause for the development of NCSE like central nervous system infections, toxins, trauma or metabolic causes. There have been reports of JME from India and the mean interval between the onset of JME and its diagnosis is very long, up to 6.8 years.[12],[13] This has been attributed to the lack of awareness of the syndrome in most of the cases and misinterpretation of EEG in some. The recognition of NCSE in patients with JME can be even more challenging since the patients may present with subtle changes in mentation. This case is presented to illustrate the fact that NCSE, even though not a very common occurrence in JME, should be kept in mind when a patient with previous history of seizures presents with subtle changes in sensorium with no obvious cause. References
Copyright 2003 - Neurology India Free full text also available from: http://www.neurologyindia.com/article.asp?issn=0028-3886;year=2003;volume=51;issue=4;spage=531;epage=533;aulast=Chemmanam The following images related to this document are available:Photo images[ni03169f1.jpg] [ni03169f2.jpg] |
| |||||||||

{kind=link}
{kind=link}