
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 52, Num. 1, 2004, pp. 36-42
|
Neurology India, Vol. 52, No. 1, January-March, 2004, pp. 36-42
Review Article
Molecular diagnosis and genetic counseling for fragile X mental retardation
Pandey UB, Phadke SR, Mittal B
Department of Medical Genetics, Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow - 226014
Correspondence Address:Department of Medical Genetics, Sanjay Gandhi
Post Graduate Institute of Medical Sciences, Lucknow - 226014 shubha@sgpgi.ac.in
Code Number: ni04011
Abstract
The fragile X syndrome is the most frequent cause of inherited mental retardation.
It is caused by a dynamic mutation: the progressive expansion of polymorphic
(CGG)n trinucleotide repeats located in the promoter region of the FMRI
gene at Xq27.3. The cloning of the FMRI gene and the elucidation of the
molecular basis of the fragile X syndrome is of great importance for the
diagnosis and understanding of this unusual type of mutation. Although
extensively studied, the mechanism behind the transition from stable normal
(CGG)n alleles to the carrier state (an unstable premutation) and from
premutation to mutation is partially understood. The clinical diagnosis
of fragile X mental retardation (FXMR) is not possible as dysmorphic features
are subtle. Molecular diagnosis by Southern Blot is the confirmatory test
that makes carrier detection and prenatal diagnosis possible. As the risk
of recurrence of FXMR is high in the family and carrier relatives, an identification
of fragile X positive children, and offering carrier detection and prenatal
diagnosis to the families is very important. It is possible by screening
mentally retarded children and adults even if there is no family history
of mental retardation or typical behavioral or physical features associated
with the fragile X phenotype. In this review we have discussed the method
for the diagnosis and counseling of the families. The complexities due
to premutation and the variable severity of manifestations in carrier females
need to be understood while counseling fragile X families.
Introduction Fragile X mental retardation (FXMR) is the commonest cause of inherited mental retardation. It is caused by progressive expansion of (CGG)n repeats, in the promoter region of the FMR-1 gene at Xq27.3.[1] FXMR primarily affects males but approximately one-third of the carrier females are also found to be affected, though the severity of mental retardation in females is less than in the males.[2] The fragile X syndrome was the first triplet repeat disorder identified and served as a prototype for several diseases caused by triplet repeat expansions in the human genome.
In 1943, Martin and Bell[3] described
a family of sex-linked mental retardation without dysmorphic features.
Although this was the first description of sex-linked familial mental retardation,
an association between sex and mental retardation had been known since
long, as institutionalized mentally retarded patients showed an excess
of males among the severely retarded patients.[4] Several
reports about many other families with idiopathic mental retardation segregating
in sex-linked fashion followed the Martin-Bell paper.[5],[6],[7] In
1969, Lubs[8] observed a marker
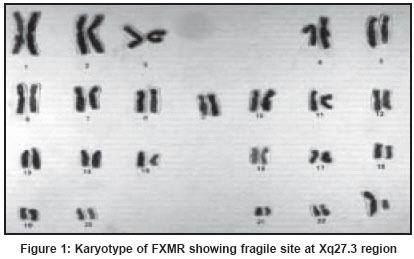
X chromosome in a family with four males affected with mental retardation.
All affected males as well as two females (one being the mother of two
affected males) expressed a constriction at the end of the long arm of
the X chromosome [Figure - 1].
It was an important and landmark observation, which led to the development
of a diagnostic method for the fragile X syndrome. In 1977, Sutherland
et al[9] showed the importance
of folic acid or thymidine deficient cell culture medium for the expression
of a fragile site on X chromosome patients. Therefore, folic acid deficient
medium remains an important condition for expressing fragile sites for
the cytogenetic diagnosis of FXMR.
During the re-examination of sex-linked mental retardation families by
Turner and Turner[10] fragile
X expression was seen in six out of sixteen families. The presence of
macroorchidism in post-pubertal affected males was also recognized as
a feature of this
syndrome. These findings were confirmed by several investigators[11],[12],[13],[14] when
several other families including the original Martin-Bell family were
re-examined. In due course of time other clinical abnormalities were
also defined. However,
there is a marked heterogeneity in the clinical presentations of FXMR.
Clinical features
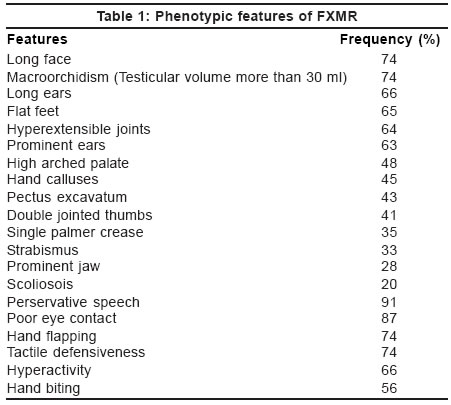
Fragile X syndrome is a very subtle dysmorphic syndrome and it is difficult to diagnose clinically [Table
- 1]. Long face with prominent mandible, large and mildly dysmorphic ears and macroorchidism are the characteristic features of fragile X syndrome. The phenotype is subtle in young children[15] and the features become prominent as the child grows. Hyperextensibilty of the finger joints, pectus excavatum, mitral valve prolapse, strabismus and epilepsy are other commonly seen features.
Mental retardation (MR) in fragile X males varies from mild to profound
with most affected males being moderately to severely retarded.[16],[17],[18] Females
are usually less severely affected than males.[18],[19],[20] A
number of behavioral characteristics associated with FXMR have been described.
They include hyperactivity, short attention span, stereotypic behavior
(hand -flapping, -rubbing, or -biting, perservative speech, echolalia),
poor eye contact, tactile defensiveness and anxiety related to social contact.[21],[22],[23],[24],[25],[26] Many
of these features suggest the possibility of autism. A combination of 11
clinical studies on fragile X males performed between 1983 and 1990, indicated
that 20% (ranging from 5 to 53%) FXMR patients had autistic
features.[27]
Due to lack of clinical diagnostic criteria, simple scoring systems have
been developed to select individuals for fragile X diagnosis[28] but
these scoring systems are mostly useful for population-based studies.
Genetics of fragile X syndrome
FXMR is an X-linked semi-dominantly inherited condition. The complex segregation pattern of the syndrome is unusual for a Mendelian trait. The occurrence of affected males and females in fragile X families suggests a dominant pattern of inheritance. However, the presence of unaffected males[29] who transmit the marker X chromosome to their daughters (also known as normal transmitting males, NTM) point to a mode of inheritance more complicated than a simple X-linked dominant mode.[3],[5],[30] Sherman et al[31],[32] performed
large-scale segregation analysis on fragile X syndrome pedigrees and observed
a significant number of asymptomatic males and affected females and put
forward a model of X-linked dominant inheritance with reduced penetrance
(79% for males and 35% for females). It was proposed that
an asymptomatic carrier male is more likely to have grandsons with the
disorder than to have brothers with FXMR. Therefore, the penetrance of
the disease increases in succeeding generations of a pedigree-an observation
now known as the Sherman paradox. The mechanism responsible for the Sherman
paradox became clear in 1991 with the cloning of the defective gene in
the fragile X syndrome.
FMR-1 gene
In order to clone the gene responsible for the fragile X syndrome, a great deal of both genetic and physical mapping was done. Although the fragile site cosegregated with the syndrome phenotype, it was not known whether the syndrome was caused by the fragile site itself or a closely linked causal mutation. Pedigree analysis localized both the causal locus and the fragile site to a 22 cM region on the X chromosome between the factor IX gene and marker St14. Further studies revealed a number of linked markers that reduced the interval to 1-2 Mb and strengthened the localization of the causative locus to the fragile site.[33]
In 1991, Fu et al[1] mapped the gene by the positional cloning method and it was designated as FMR-1 (Fragile X Mental Retardation gene 1). This gene consists of 17 exons, spanning 38 kb of Xq27.3.[29] The FMR-1 mRNA is -4.0 kb long, of which 1.9 kb is coding sequence, predicting a protein product of 631 amino acids.[34] In the non-coding part of the gene at the 5′ end, there is a section of DNA composed mainly of tandem repetitive triplets of CGG, which corresponds to the fragile site on the X chromosome.
Molecular basis of the fragile X syndrome
Almost all cases of FXMR are caused by the expansion of CGG repeats in
the 5′untranslated region of the FMR-1 gene[29] [Figure
- 2]. Normal individuals have 6 to 50 CGG repeats, which are stably transmitted from generation to generation. In fragile X families the CGG repeat number exceeds the normal range in NTM and affected individuals. According to the size and methylation status of the expansion, fragile X mutations have been classified into premutations with small expansions of 50-200 copies of the CGG repeat (unmethylated), and full mutations ranging in size above 200 repeats, with concomitant hypermethylation of the nearby CpG island and the expanded CGG repeat itself. Full mutations are detected in individuals affected by the fragile X syndrome and also in a proportion of unaffected carrier females. As yet there is no universal agreement as to the number of repeats that should define the lower limit for the premutation range although most screening studies have used 55 repeats.
In contrast to the normal FMR-1 gene, premutations are unstable. Their
instability is seen in virtually every transmission when passing to the
offspring, size increases being much more common than occasional contractions
of the repeat. One important factor that influences the rate and size of
an expansion is the gender of the parent transmitting the unstable CGG
repeat. A premutation has been documented to expand to a full mutation
in offspring with concomitant abnormal methylation only when it is transmitted
by a female.[19] The size
of the CGG repeat expansion is another factor having a significant effect
on instability; the larger the premutation, the more unstable it is upon
transmission. Moreover, the risk of transition of a premutation to a full
mutation, and consequently the risk of having an affected child has been
shown to depend strongly on the size of the maternal permutation.[1],[36] It
remains to be explained how an expansion of the (CGG)n repeats leads to
hypermethylation of the CpG islands.
Recent studies have completely changed the preexisting ideas about the
premutation alleles of FMR-1 that other than serving as a source for full-mutation
alleles in matrilineal transmissions of fragile X syndrome, such alleles
do not give rise to clinical involvement. It is now clear that premutation
alleles also contribute directly to clinical involvement. Although premutation
females were reported to have normal cognitive abilities[35] premature
ovarian failure has been observed in 21% of such carriers.[19] Mild
emotional problems have also been reported in 20% of the carrier females
that are correlated with the number of CGG repeats.[37] Recent
long-term follow-up studies show neurological signs involving intention tremor,
ataxia, and cognitive decline, particularly among older male carriers of
premutation alleles of the FMR-1 gene.[38] Premutation
males may occasionally have cognitive involvement in childhood,[39] and
a subgroup is at risk for late-onset neurological problems including executive
function deficits, tremor, ataxia, and brain atrophy.[38] It
is yet to be determined why certain subgroups of males and females with premutation
are more vulnerable to CNS problems.
A small group of patients with fragile X phenotype do not show expansion
of CGG repeats.[40] Intragenic
deletion or point mutations in the FMR-1 gene have been observed in these
patients.[41],[42],[43],[44],[45],[46]
Stability of CGG repeats
In the normal size range the CGG trinucleotide repeat alleles behave like other microsatellite markers, and they are stable upon transmission.[1] DNA sequencing of normal and premutation FMR-1 alleles has revealed that the number and position of AGG interruptions within the CGG repeat sequence may significantly influence the stability of the repeat, and in particular, the length of an uninterrupted CGG repeat sequence appears to be an important determinant of instability. Most normal alleles contain one or more regularly spaced AGG units and the uninterrupted CGG tracts do not exceed 30 repeats. In contrast, premutation alleles typically have none or at the most one interspersing AGG triplet and a CGG copy number exceeding 30 repeats at the 3′ end of the repeat array. The AGG interspersions appear to confer stability and their absence gives rise to longer perfect CGG arrays with increased instability and predisposition to expansion.[47] Nolin et al[48] have
further suggested that in addition to the repeat length and the AGG content,
as yet unidentified familial factors might influence the stability of CGG
repeats.
The CGG repeat alleles that expand to pre- and full mutations
have been shown to be in linkage disequilibrium with microsatellite[49] within
the FMR-1 gene or close to its 5′ end. A significant linkage disequilibrium between the fragile X syndrome and certain microsatellite haplotypes suggests that fragile X mutations arose several thousands of years back and most of the present fragile X chromosomes have ancient origins.[49] This
hypothesis is supported by the studies done in a genetically isolated Finnish
population where 80% fragile X chromosomes and 8% of the normal chromosomes were found to be associated with certain haplotypes.[50] A
similar kind of linkage disequilibrium was also observed in other Caucasian
and African populations.[49] However,
the disequilibrium has not been established in Asian populations including
India.
Fragile X mental retardation protein (FMRP)
FMRP is a RNA-binding protein of 68-70 KD, in which extensive alternative splicing occurs. It is widely expressed with particularly high levels in the brain and testis.[51] Postmortem examination of the brains of fragile X patients and FMRP-deficient mice showed very long and thin dendritic spines in the neocortex. A unifying model for FMRP function is that it shuttles specific mRNAs from the nucleus to postsynaptic sites where mRNAs are held in a transitionally inactive form until synaptic input changes FMRP activity to allow mRNA translation. Recently, it has been observed that FMRP is associated with large numbers of mRNA whose genes are involved in important neuronal functions such as vesicle transport, signal transduction, etc. In the absence of FMRP these mRNAs become misregulated which may in turn lead to the mental retardation.[52]
Prevalence of the fragile X syndrome
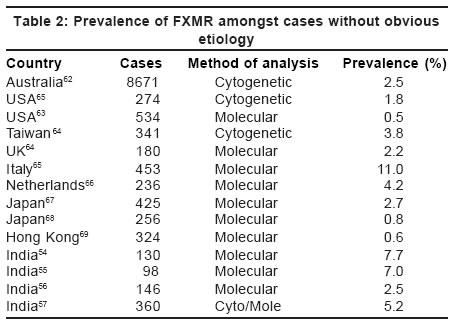
FXMR has been detected in all populations and ethnic groups studied with different
frequencies. Most of the studies show prevalence of FXMR amongst the target
population of mentally retarded males of unknown etiology between 0.5 to 3%.[53] However,
higher prevalence up to 11% has also been reported [Table
- 2].
It may vary from population to population but also depends on the selection
of cases for study. Indian studies from New Delhi and Kolkata show a prevalence
of 7 and 7.5% respectively amongst the mentally retarded population.[54],[55] Our
experience of the molecular screening of 146 mentally retarded males without
obvious etiology showed 2.5% prevalence of FXMR.[56] Cytogenetic analysis of individuals with mental retardation showed the prevalence of fragile X as 1/1200-1/2600 in males and 1/1600-1/4200 in females.[58],[59],[60] This was probably an overestimation. Molecular techniques estimate the prevalence of FXMR as ranging from 1/3717-1/8918 in the Caucasian male population.[61] For females, recent large studies have established the high prevalence of premutation carriers with a range from 1/246-1/468 in the general population.[61]
Diagnosis of fragile X mental retardation
Cytogenetic diagnosis of fragile X became possible when Sutherland showed that folic acid deficient cell culture medium could induce a chromosomal fragile site at Xg27.3, which was found to be linked to the mental retardation. After the mapping of the FMR-1 gene, cytogenetic study is no longer considered a diagnostic method because of its false positive and false negative results. Major progress in molecular diagnosis was made soon after the cloning of the FMR-1 gene and a direct molecular test became available which is confirmatory for fragile X diagnosis. In addition to direct mutation analysis, it is now possible to demonstrate the expression of FMR-1 at the protein level by using monoclonal antibodies directed against FMRP.[70]
Southern analysis
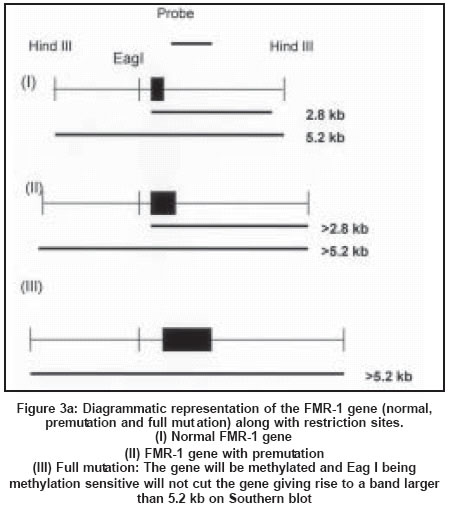
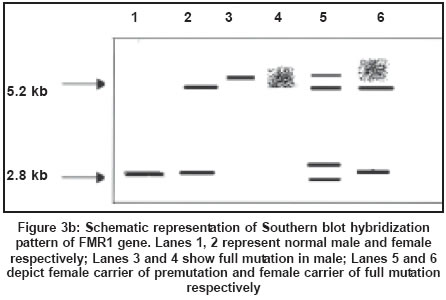
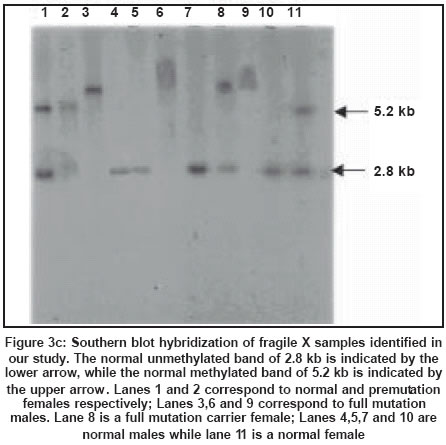
Southern blot analysis is considered as the gold standard for fragile X diagnosis.
It can clearly distinguish between mutation and premutation alleles and can
also provide information regarding methylation status. Digestion of genomic
DNA from the patients with restriction enzymes spanning the FMRI (CGG)n region
followed by Southern hybridization with a radioactive probe is now the preferred
method for the diagnosis of fragile X syndrome[71],[72] [Figure
- 3 a, b, c]. A double digest using EcoRI and the methylation sensitive enzyme
Eagl or BssHII performs methylation studies of fragile X chromosomes. Since
FMR-l
is almost always methylated when the CGG expansion is beyond 230 repeats in
males with full mutation, there is an increase in the size of the band corresponding
to the FMR-1 gene[72] [Figure
- 3 a, b, c].
One of the two X chromosomes in a normal female is inactivated and the FMR-1
gene is methylated as a result of the process of lyonisation. An inactive
X chromosome with a normal FMR-1 gene shows a 5.2 kb band while an active X
with a normal FMR-1 gene in females and in males shows a 2.8 kb band. In the
presence of premutation, the 2.8 kb PCR product increases in size. A female
carrier with full mutation shows three bands corresponding to the normal unmethylated
female pattern (active state - 2.8 Kb), methylated (inactive state - 5.2 kb)
and an abnormal band of size greater than 5.2 kb reflecting hypermethylation
and expansion of the FMR-1 mutation. Full mutations are highly unstable and
give rise to smearing of the band. Mosaics for full mutations and premutation
could be detected by the Southern hybridization method. Southern blot hybridization
is time-consuming, costly and labor intensive. The limitation of Southern blot
is that it cannot give the exact number of repeats which is especially necessary
for premutation carriers and alleles in the gray zone, i.e. between 45 to 55
repeats.
Polymerase chain reaction (PCR)
Due to high guanine cytosine (GC) content,
amplification of CGG repeat-containing regions becomes difficult by PCR. However,
now the expansion of CGG repeat can be detected by using modified PCR methods.
The advantages of PCR are faster diagnosis, requirement of only a small amount
of DNA (<100 ng) and accurate sizing of the trinucleotide repeat in the FMR-l gene. PCR-based diagnosis is feasible and reliable in the premutation (60-100 CGG repeats) carriers also. A lot of modifications have been made in the PCR protocol to increase the probability of amplifying across longer alleles. The use of nucleotide analog 7-deaza guanosine triphosphate makes amplification of long GC-rich repeats possible.
The disadvantages of PCR are that it is difficult to detect full mutation
alleles because of technical difficulties in performing PCR across hundreds
of tandem repeated CGG triplets where the high content of GC and strong secondary
structure make the amplification difficult. PCR also cannot detect mosaicism
between premutation and normal alleles due to differential amplification. For
many PCR protocols, the DNA fragment with the expanded repeats does not amplify.
This is especially problematic for females and persons with repeat size mosaicism
who could be misdiagnosed as normal. PCR based methods can be used for screening
for Fragile X syndrome. The samples which fail to amplify by PCR and any female
who appears to be homozygous should be tested by Southern blot analysis.
Antibody test
The antibody-based diagnostic method for detecting the presence or absence of FMRP is also possible in lymphocytes.[70] Cells of fragile X males with methylated full mutation produce no FMRP, while in individuals with normal FMR-1 or in premutation carriers, FMRP can be detected in the cytoplasm of lymphocytes. The test can be used to diagnose affected males but cannot be used to identify female carriers of full mutation, as FMRP is still produced by the normal X chromosome.[70] In addition, the antibody test is unable to differentiate between normal and premutation alleles. Thus, at present its reliable use is limited to population screening for the diagnosis of affected males. Genetic counseling
Associated mental impairment and high risk of recurrence makes genetic counseling
essential for families with fragile X syndrome. With the availability of molecular
tests, carrier detection and prenatal diagnosis is now possible. The most important
step is the diagnosis of the affected child. As clinical diagnosis is not possible,
especially in young children, it is necessary to test all mentally retarded
children without any obvious etiology. Like any X-linked disorder the chances
that the son or daughter will inherit the mutated chromosome from the mother
are 50%. However, whether the child will be affected or unaffected will depend on whether the FMR-1 gene harbors full mutation or premutation. If the mother is the carrier of full mutation, her 50% sons and 50% daughters will inherit the mutation. Those sons will be clinically affected, but prediction of the clinical phenotype and severity is not possible in females.
If the mother is a carrier of premutation, the chance that the premutation
will be converted to full mutation varies accordingly as the number of CGG
repeats generally increase in the mother. If the number of CGG repeats in the
mother is 60 to 80 or 80 to 100, then the chances that permutation will get
converted to full mutation are 14% to 55% and 80% to 90% respectively. If the number of repeats in the mother is 100 to 200 the chance of it getting expanded to full mutation is almost 100%.[73]
Molecular diagnosis can provide a reliable prenatal diagnosis. But as mentioned
above, the clinical phenotype of the female carriers of full mutation can vary
from normal to significant mental retardation and cannot be predicted prenatally.
As this condition is X-linked, screening of female relatives of the mother
for identification of carriers is useful. An attempt should be made to educate
the family, to discuss with their relatives the necessity of carrier detection
and genetic counseling to prevent recurrence of similarly affected children
in the family.[74]
FXMR being one of the leading causes of mental retardation, screening of
the general population for identification of carrier females and offering
them
prenatal diagnosis for prevention of the birth of a mentally handicapped
child, has also been tried and found feasible in the developed countries.[75] Though
this approach is not presently feasible in India, increased awareness amongst
the clinicians about fragile X syndrome as a cause of mental retardation,
is necessary. DNA tests for fragile X syndrome of all mentally retarded children
without an obvious cause, and genetic counseling of the families will greatly
help in reducing the burden on the families.
At present, there is no cure for the fragile X syndrome. A wide variety of
therapeutic measures are used to take care of the special educational needs
of the individuals with FXMR and to make them as independent as possible.
Pharmacological agents like antidepressants, anxiety medications and anticonvulsants
can be
used as per indications. The aim of management is to help the children
and adults with FXMR learn to function in the household environment and be
employed
in constructive occupations in sheltered atmospheres. Speech therapy may
be needed. In addition, support from the family members, community and
non-governmental organizations is needed for better management of these unfortunate
victims
of inherited mental retardation.
References
| 1. | Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991;67:1047-58. Back to cited text no. 1 [PUBMED] |
| 2. | Rousseau F. The fragile X syndrome: implications of molecular genetics for the clinical syndrome Eur J Clin Invest 1994;24:1-10. Back to cited text no. 2 |
| 3. | Martin JB, Bell J. A pedigree of mental defect showing sex-linkage. J Neurol Psychiatr 1943;6:154-7. Back to cited text no. 3 |
| 4. | Penrose LS. A clinical and genetic study of 1280 cases of mental defect: special report series No. 229. London: Medical Research Council; 1938. Back to cited text no. 4 |
| 5. | Losowsky MS. Hereditary mental defect showing the pattern of sex influence. J Ment Defic Res 1961;5:60-2. Back to cited text no. 5 [PUBMED] |
| 6. | Renpenning H, Gerrard JW, Zaleski WA, Tabata T. Familial sex-linked mental retardation. Canad. Med Ass J 1962;87:954-6. Back to cited text no. 6 [PUBMED] |
| 7. | Dunn HG, Renpenning H, Gerrard JW. Mental retardation as a sex-linked defect. Am J Ment. Defic 1963;67:827-48. Back to cited text no. 7 |
| 8. | Lubs HA. A marker X chromosome. Am J Hum Genet 1969;21:231-44. Back to cited text no. 8 [PUBMED] |
| 9. | Sutherland GR. Fragile sites on human chromosomes: demonstration of their dependence on the type of tissue culture medium. Science 1977;197:265-6. Back to cited text no. 9 [PUBMED] |
| 10. | Turner G, Turner B. X-linked mental retardation. J Med Genet 1974;11:109-13. Back to cited text no. 10 [PUBMED] |
| 11. | Jacobs PA, Glover TW, Mayer M, Fox P, Gerrard JW, Dunn HG, et al. X-linked mental retardation: a study of 7 families. Am J Med Genet 1980;7:471-89. Back to cited text no. 11 [PUBMED] |
| 12. | Jennings M, Hall JG, Hoehn H. Significance of phenotypic and chromosomal abnormalities in X-linked mental retardation (Martin-Bell or Renpenning syndrome). Am J Med Genet 1980;7:417-32. Back to cited text no. 12 [PUBMED] |
| 13. | Martin RH, Lin CC, Mathies BJ, Lowry RB. X-linked mental retardation with macro-orchidism and marker-X chromosomes. Am J Med Genet 1980;7: Back to cited text no. 13 |
| 14. | 433-41. Back to cited text no. 14 |
| 15. | Richards BW, Sylvester PE, Brooker C. Fragile X-linked mental retardation: the MartinBell syndrome. J Ment Defic Res 1981;25:253-6. Back to cited text no. 15 |
| 16. | Lachiewicz AM, Dawson VV, Spiridigliozzi UA. Physical characteristics of young boys with fragile X syndrome: reasons for difficulties in making a diagnosis in young males. Am J Med Genet 2000;92:229-36. Back to cited text no. 16 |
| 17. | Pennington BF, O'Connor RA, Sudhalter V. Towards a neuropsychology of fragile X syndrome. In: Hagerman R.J. and Silverman A.C editors. Fragile X syndrome: diagnosis, treatment and research. The Johns Hopkins University Press Ltd, London 1991. pp. 173-201. Back to cited text no. 17 |
| 18. | Curfs LM, Wiegers AM, Fryns JP. Intelligence and the fra (X) syndrome: a review. Genet Couns 1991;2:55-62. Back to cited text no. 18 |
| 19. | Oostra BA, Willemsen R. The X chromosome and fragile X mental retardation. Cytogenet Genome Res 2002;99:257-64. Back to cited text no. 19 |
| 20. | Rousseau F, Heitz D, Oberle I, Mandel JL. Selection in blood cells from female carriers of the fragile X syndrome: inverse correlation between age and proportion of active X chromosomes carrying the full mutation. J Med Genet 1991;8:830-6. Back to cited text no. 20 |
| 21. | de Vries BB, Wiegers AM, Smits AP, Mohkamsing S, Duivenvoorden HJ, Fryns JP, et al. Mental status of females with an FMR1 gene full mutation. Am J Hum Genet 1996;58:1025-32. Back to cited text no. 21 |
| 22. | Brown WT, Jenkins EC, Friedman E, Brooks J, Wisniewski K, Raguthu S, et al. Autism is associated with the fragile-X syndrome. J Autism Dev Disord 1982;12:303-8. Back to cited text no. 22 |
| 23. | Hagerman RJ, Jackson AW, Levitas A, Rimland B, Braden M. An analysis of autism in fifty males with the fragile X syndrome. Am J Med Genet 1986;23:359-74. Back to cited text no. 23 |
| 24. | Reiss AL, Feinstein C, Toomey KE, Goldsmith B, Rosenbaum K, Caruso MA. Psychiatric disability associated with the fragile X chromosome. Am J Med Genet 1986;23:393-401. Back to cited text no. 24 |
| 25. | Hagerman RJ, Sobesky WE. Psychopathology in fragile X syndrome. Am J Orthopsychiatry 1989;59:142-52. Back to cited text no. 25 |
| 26. | Hagerman RJ. Physical and behavioral phenotype. In: Hagerman RJ, Silverman MS, editors. Fragile X syndrome. Diagnosis treatment and research. London: Johns Hopkins Press Ltd; 1991. pp. 3-68 Back to cited text no. 26 |
| 27. | Reiss AL, Freund L. Behavioral phenotype of fragile X syndrome: DSM-III-R autistic behavior in male children. Am J Med Genet 1992;43:35-46. Back to cited text no. 27 |
| 28. | Hagerman RJ, Silverman AC. Fragile X syndrome: Diagnosis, treatment and research. Baltimore: Johns Hopkins Press Ltd; 1991. Back to cited text no. 28 |
| 29. | Hagerman RJ, Amiri K, Cronister A. Fragile X checklist. Am J Med Genet 1991;38:283-7. Back to cited text no. 29 |
| 30. | Fryns JP, van den Berghe H. Transmission of fragile (X)(827) from normal male(s). Hum Genet 1982;61:262-3. Back to cited text no. 30 |
| 31. | Nielsen KB, Tommerup N, Poulsen H, Mikkelsen M. X-linked mental retardation with fragile X. A pedigree showing transmission by apparently unaffected males and partial expression in female carriers. Hum Genet 1981;59:23-5. Back to cited text no. 31 |
| 32. | Sherman SL, Morton NE, Jacobs PA, Turner G. The marker (X) syndrome: a cytogenetic and genetic analysis. Ann Hum Genet 1984;48:21-37. Back to cited text no. 32 |
| 33. | Sherman SL, Jacobs P, Morton N, Froster-Iskenius U, Howard-Peebles P, Nielsen K, et al. Further segregation analysis of the fragile X syndrome with special reference to transmitting males. Hum Genet 1985;69:289-99. Back to cited text no. 33 |
| 34. | Oberlé I, Heilig R, Moisan J, Kloepher C, Mattei M, Mattei J, et al. Genetic analysis of the fragile-X mental retardation syndrome with two flanking polymorphic DNA-markers. Proc Natl Acad Sci USA 1986;83:1016-20. Back to cited text no. 34 |
| 35. | Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991;65:905-14. Back to cited text no. 35 |
| 36. | Hagerman RJ, Hagerman PJ. The fragile X premutation: into the phenotypic fold. Curr Opin Genet Dev 2002;12:278-83 Back to cited text no. 36 |
| 37. | Heitz D, Devys D, Imbert G, Kretz C, Mandel JL. Inheritance of the fragile X syndrome: size of the fragile X premutation is a major determinant of the transition to full mutation. J Med Genet 1992;29:794-801. Back to cited text no. 37 |
| 38. | Johnston C, Eliez S, Dyer-Friedman J, Hessl D, Glaser B, Blasey C, et al. Neurobehavioral phenotype in carriers of the fragile X premutation. Am J Med Genet 2001;103:314-9. Back to cited text no. 38 |
| 39. | Hagerman PJ, Greco CM, Hagerman RJ. A cerebellar tremor/ ataxia syndrome among fragile X premutation carriers. Cytogenet Genome Res 2003;100:206-12 Back to cited text no. 39 |
| 40. | Tassone F, Hagerman RJ, Chamberlain WD, Hagerman PJ. Transcription of the FMR1 gene in individuals with fragile X syndrome. Am J Med Genet 2000;97:195-203. Back to cited text no. 40 |
| 41. | Mila M, Castellvi-Bel S, Sanchez A, Barcelo A, Badenas C, Mallolas J, et al. Rare variants in the promoter of the fragile X syndrome gene (FMR1). Mol Cell Probes 2000;14:115-9. Back to cited text no. 41 |
| 42. | Gedeon AK, Baker E, Robinson H, Partington MW, Gross B, Manca A, et al. Fragile X syndrome without CCG amplification has an FMRI deletion. Nat Genet 1992;1:341-4. Back to cited text no. 42 |
| 43. | Wohrle D, Kotzot D, Hirst MC, Manca A, Korn B, Schmidt A, et al. A microdeletion of less than 250 kb, including the proximal part of the FMR-l gene and the fragile-X site, in a male with the clinical phenotype of fragile-X syndrome. Am J Hum Genet 1992;51:299-306. Back to cited text no. 43 |
| 44. | Tarleton J, Richie R, Schwartz C, Rao K, Aylsworth AS, Lachiewicz A. An extensive de novo deletion removing FMRI in a patient with mental retardation and the fragile X syndrome phenotype. Hum Mol Genet 1993;2:1973-4. Back to cited text no. 44 |
| 45. | Gu Y, Lugenbeel KA, Vockley JG, Grody WW, Nelson DL. A de novo deletion in FMRI in a patient with developmental delay. Hum Mol Genet 1994;3:1705-6. Back to cited text no. 45 |
| 46. | Quan F, Grompe M, Jakobs P, Popovich BW. Spontaneous deletion in the FMRI gene in a patient with fragile X syndrome and cherubism. Hum Mol Genet 1995;4:16814. Back to cited text no. 46 |
| 47. | Quan F, Zonana J, Gunter K, Peterson KL, Magenis RE, Popovich BW. An atypical case of fragile X syndrome caused by a deletion that includes the FMRI gene. Am J Hum Genet 1995;56:1042-51. Back to cited text no. 47 |
| 48. | Kunst CB, Zerylnick C, Karickhoff L, Eichler E, Bullard J, Chalifoux M, et al. FMR1 in global populations. Am J Hum Genet 1996;58:513-22. Back to cited text no. 48 |
| 49. | Nolin SL, Lewis FA, Ye LL, Houck GE, Glicksman AE, Limprasert P, et al. Familial transmission of the FMR1 CGG repeat.Am J Hum Genet 1996;59:1252-61. Back to cited text no. 49 |
| 50. | Chiurazzi P, Macpherson J, Sherman S, Neri G. Significance of linkage disequilibrium between the fragile X locus and its flanking markers. Am J Med Genet 1996;64:203-8. Back to cited text no. 50 |
| 51. | Haataja R, Vaisanen ML, Li M, Ryynanen M, Leisti J. The fragile X syndrome in Finland: demonstration of a founder effect by analysis of microsatellite haplotypes. Hum Genet 1994;94:479-83. Back to cited text no. 51 |
| 52. | Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet 1993;4:335-40. Back to cited text no. 52 |
| 53. | Mittal B, Pandey UB. New insights into the function of fragile X mental retardation protein. Hotspot. Clin Genet 2002;62:191-5. Back to cited text no. 53 |
| 54. | Crawford DC, Acuna JM, Sherman SL. FMRI and the fragile X syndrome: human genome epidemiology review. Genet Med 2001;3:359-71. Back to cited text no. 54 |
| 55. | Sharma D, Gupta M, Thelma BK. Expansion mutation frequency and CGG/GCC repeat polymorphism in FMR1 and FMR2 genes in an Indian population. Genet Epidemiol 2001;20:129-44. Back to cited text no. 55 |
| 56. | Saha S, Karmakar P, Chatterjee C, Banerjee D, Das S, Dasgupta UB. Fragile X syndrome in Calcutta, India. Ann Clin Biochem 2001;38:264-71. Back to cited text no. 56 |
| 57. | Pandey UB, Phadke S, Mittal B. Molecular screening of FRAXA and FRAXE in Indian patients with unexplained mental retardation. Genet Test 2002;6:335-9 Back to cited text no. 57 |
| 58. | Jain U, Verma IC, Kapoor AK. Prevalence of fragile X(A) syndrome in mentally retarded children at a genetics referral centre in Delhi, India. Ind J Med Res 1998;108:12-6. Back to cited text no. 58 |
| 59. | Turner G, Robinson H, Laing S, Purvis-Smith S. Preventive screening for the fragile X syndrome. N Engl J Med 1986;315:607-9. Back to cited text no. 59 |
| 60. | Webb TP, Bundey SE, Thake AI, Todd J. Population incidence and segregation ratios in the Martin-Bell syndrome. Am J Med Genet 1986;23:573-80 Back to cited text no. 60 |
| 61. | Kahkonen M, Alitalo T, Airaksinen E, Matilainen R, Launiala K, Autio S, et al. Prevalence of the fragile X syndrome in four birth cohorts of children of school age. Hum Genet 1987;77:85-7. Back to cited text no. 61 |
| 62. | Turner G, Robinson H, Ling S, van der Berk M, Colley A, Goddard A, et al. Population screening for fragile X. Lancet 1992:339;1210-3. Back to cited text no. 62 |
| 63. | Jacobs PA, Mayer M, Matsuura J, Rhoads F, Yee SC. A cytogenetic study of a population of mentally retarded males with special reference to the marker (X) syndrome. Hum Genet 1983;63:139-48. Back to cited text no. 63 |
| 64. | Li SY, Tsai CC, Chou MY, Lin JK. A cytogenetic study of mentally retarded school children in Taiwan with special reference to the fragile X chromosome. Hum Genet 1988;79:292-6. Back to cited text no. 64 |
| 65. | Jacobs PA, Bullman H, Macpherson J, Youings S, Rooney V, Watson A, et al. Population studies of the fragile X: a molecular approach. J Med Genet 1993;30:454-9. Back to cited text no. 65 |
| 66. | Perroni L, Grasso M, Argusti A, Lo Nigro C, Croci GF, Zelante L, et al. Molecular and cytogenetic analysis of the fragile X syndrome in a series of 453 mentally retarded subjects: a study of 87 families. Am J Med Genet 1996;64:176-80. Back to cited text no. 66 |
| 67. | van den Ouweland AM, de Vries BB, Bakker PL, Deelen WH, de Graaff E, et al. DNA diagnosis of the fragile X syndrome in a series of 236 mentally retarded subjects and evidence for reversal of mutation in the FMR-1 gene Am J Med Genet 1994;51:482-5. Back to cited text no. 67 |
| 68. | Hofstee Y, Arinami T, Hamaguchi H. Comparision between the Cytogenetic test for fragile X and the molecular analysis in Japanese mentally retarded individuals Am J Med Genet 1994:51;466-70. Back to cited text no. 68 |
| 69. | Nanba E, Kohno Y, Matsuda A, Yano M, Sato C, Hashimoto K, et al. Non-radioactive DNA diagnosis for the fragile X syndrome in mentally retarded Japanese males. Brain Devel 1995;17:317-21. Back to cited text no. 69 |
| 70. | Pang CP, Poon PM, Chen QL, Lai KY, Yin CH, Zhao Z, et al. Trinucleotide CGG repeat in the FMRI gene in Chinese mentally retarded patients. Am J Med Genet 1999;84:179-83. Back to cited text no. 70 |
| 71. | Willemsen R, Mohkamsing S, de Vries B, Devys D, van den Ouweland A, Mandel JL, et al. Rapid antibody test for fragile X syndrome. Lancet 1995;345:1147-8. Back to cited text no. 71 |
| 72. | Oostra BA, Jacky PB, Brown WT, Rousseau F. Guidelines for the diagnosis of fragile X syndrome. National Fragile X Foundation. J Med Genet 1993;30:410-3. Back to cited text no. 72 |
| 73. | Warren ST, Nelson DL. Advances in molecular analysis of fragile X syndrome. JAMA 1994;271:536-42. Back to cited text no. 73 |
| 74. | Nolin SL, Brown WT, Glicksman A, Houck GE, Gargano AD, Sullivan A, Biancalana V, et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet 2003;72:454-64. Back to cited text no. 74 |
| 75. | McConkie-Rosell A, Robinson H, Wake S, Staley LW, Heller K, Cronister A. Dissemination of genetic risk information to relatives in the fragile X syndrome: guidelines for genetic counselors Am J Med Genet 1995;59:426-30. Back to cited text no. 75 |
| 76. | Ryynanen M, Heinonen S, Makkonen M, Kajanoja E, Mannermaa A, Pertti K. Feasibility and acceptance of screening for fragile X mutations in low-risk pregnancies. Eur J Hum Genet 1999;7:212-6. Back to cited text no. 76 [PUBMED] [FULLTEXT] |
Copyright 2004 - Neurology India
The following images related to this document are available:
Photo images
[ni04011f3b.jpg]
[ni04011f3c.jpg]
[ni04011f3a.jpg]
[ni04011f2.jpg]
[ni04011f1.jpg]
[ni04011t2.jpg]
[ni04011t1.jpg]
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}