
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 52, Num. 2, 2004, pp. 210-212
|
Neurology India, Vol. 52, No. 2, April-June, 2004, pp. 210-212
Original Article
Spectrum of epilepsy in tuberous sclerosis
Anisya-Vasanth AV, Satishchandra P, Nagaraja D, Swamy HS, Jayakumar PN
Departments of Neurology and Neuroiamging, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore
Correspondence Address:Departments of Neurology and Neuroiamging, National Institute of Mental Health and Neurosciences (NIMHANS), Bangalore
anisyavasanth@yahoo.com
Code Number: ni04064
Abstract
Tuberous sclerosis (TS) is an autosomal dominant disease that affects the brain, skin, eye, heart and kidney. The diagnostic criteria for tuberous sclerosis complex (TSC) have recently been revised. There are relatively few Indian studies on this disorder. Twenty-six patients diagnosed as having TS over a period of 18 years are being reported. The onset of seizures ranged from infancy to adolescence. The patterns of epilepsy encountered were generalized tonic clonic seizures (13), complex partial seizures (10), simple partial seizures (9) and myoclonic jerks (4) including infantile spasms (3). Patients often had more than one seizure type. Nineteen patients were mentally subnormal. Cutaneous manifestations were facial angiofibroma i.e. adenoma sebaceum (20), shagreen patches (7), hypopigmented macules (6), ash leaf spots (4), café-au-lait spots (2), facial hypoplasia (2) and periungual fibromas (1). One patient each had retinal phakoma and renal angiomyolipoma. CT scan revealed sub-ependymal calcifications (12), parenchymal tubers (3), cerebral edema (3) and cortical atrophy (1). One patient had enhancement of peri-ventricular sub-ependymal lesions on MRI. Anticonvulsants prescribed were phenobarbitone (20), diphenyl hydantoin (14), carbamazepine (8), sodium valproate (4), benzodiazepines (4), ACTH (2), prednisone (1), mysoline (1) and vigabatrin (1). Most patients were on combinations of anti-convulsants and response to therapy was usually not very satisfactory. However, the child treated with vigabatrin did well.
Keywords: Epilepsy, facial angiofibroma, hypsarrhythmia,
hamartin, sub-ependymal nodules, tuberin, tuberous sclerosis,
vigabatrin
Introduction Tuberous sclerosis (TS) is an autosomal dominant disorder that affects the brain, skin, eye, heart and kidney. Neuro-behavioral features include epilepsy, mental subnormality and pervasive developmental disorder. Manifestations of TS outside the central nervous system are characterized by hypomelanotic skin macules, facial angiofibromas (adenoma sebaceum), ungual fibromas, renal angiomyolipomas and cardiac rhabdomyomas. There have been relatively few publications on TS from India.[1],[2] Tuberous sclerosis complex (TSC) results from mutations in one of two recently identified genes, TSC1 on chromosome 9q34 and TSC2 on chromosome 16p13, that encode distinct proteins, hamartin and tuberin respectively.[3],[4],[5] TSC1 mutations are slightly less common in sporadic TSC cases (no family history, approximately two-thirds of all patients) and are more common in familial cases.[6] Recent advances in molecular genetics and the favorable response of epilepsy in TS to therapy with vigabatrin have rekindled interest in this neuro-cutaneous syndrome. Material and methods
The case records of patients diagnosed as having TS at NIMHANS over a period of 18 years (1982-2000) were analyzed with reference to the age of onset of symptoms, type of epilepsy, mental subnormality, family history, radiological features, nature of medication and response to therapy.Results Twenty-six patients were diagnosed as having TS during this 18-year period (M:F=16:10). Ten of them presented in infancy and childhood while 16 presented in adulthood. The onset of seizures was in infancy in 9 (infantile spasms-3), childhood in 11 and adolescence in 6.
The patterns of epilepsy encountered were: generalized tonic clonic seizures-13, complex partial seizures-10, simple partial seizures-9 and myoclonic jerks-4 including infantile spasms-3. Patients often had more than one seizure type. Mental subnormality was encountered in 19 patients, 10 of whom presented in infancy and childhood. Six patients, all with childhood onset, had hyperkinetic behavior. Two adults also had behavioral problems and one adult had left hemiplegia.
Various cutaneous manifestations encountered were as follows: facial angiofibroma (adenoma sebaceum)-20, shagreen patches-7, hypopigmented macules-6, ash leaf spots-4, café-au-lait spots-2, facial hypoplasia-2 and periungual fibromas-1. One patient each was detected to have retinal phakoma and renal angiomyolipoma. Family history of epilepsy was present in 3 children. Fathers of 2 children were detected to have hypopigmented macules and facial angiofibroma. However, they were asymptomatic neurologically.
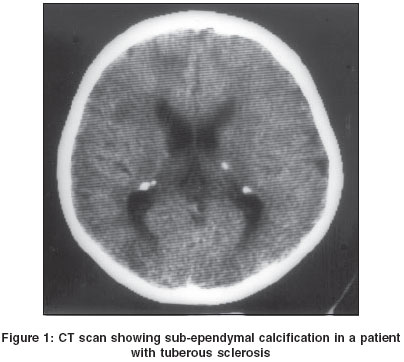
CT scan revealed sub-ependymal calcifications-12 [Figure - 1], parenchymal tubers-3, cerebral edema-3, cortical atrophy-1 and enhancement of periventricular sub-ependymal lesions on MRI in one patient. Electro-encephalography recording was carried out in 14 patients and revealed generalized seizure discharges in 5, focal seizure discharges in 5 and hypsarrhythmia in 3.
Anticonvulsants prescribed were: phenobarbitone-20, diphenylhydantoin-14, carbamazepine-8, sodium valproate-4, benzodiazepines-4, ACTH-2, prednisone-1, mysoline-1 and vigabatrin-1. Most patients were on combinations of anticonvulsants and response to therapy was usually not very satisfactory. A two-and-a-half-year-old boy with delayed milestones, shagreen patch, ash leaf spots and facial angiofibroma had recurrent myoclonic jerks poorly controlled with ACTH and valproate. There was significant improvement after adding vigabatrin and at follow-up two-and-a-half years later he was free from seizures with normal ophthalmic evaluation. Discussion The diagnostic criteria for TSC were revised recently.[7] Accordingly, the clinical and radiological features of TSC have now been divided into major and minor categories based on the apparent degree of specificity for TSC of each feature. A definite diagnosis of TSC now requires two or more distinct types of lesions rather than multiple lesions of the same type in the same organ system.[7]
The major features proposed are tubers, sub-ependymal nodules (SEN), sub-ependymal giant cell astrocytomas (SEGAS), cardiac rhabdomyoma, retinal hamartomas, renal angiomyolipomas, pulmonary lymphangiomyomatosis, shagreen patch, facial angiofibroma (adenoma sebaceum) and ungual fibromas. The minor features proposed are dental pitting, bone cysts, hypomelanotic macules, pulmonary lymphangiomyomatosis, gingival fibromas, hamartomatous rectal polyps and cerebral white matter radiation lines on MRI. Definite TSC is diagnosed in the presence of two major features or one major plus two minor features, probable TSC with one major and one minor feature and possible TSC with either one major or two minor features.
Neuropsychiatric disorders are very frequently encountered in TSC. Epilepsy may develop in infancy as infantile spasms, but may also arise in childhood or adulthood as complex partial seizures, myoclonic jerks, generalized tonic clonic seizures and absences. Curatolo et al[8] described clinical and EEG differences between infantile spasms in TSC and the classical West syndrome. In TSC, seizures at onset are partial motor seizures and infantile spasms. Subtle partial seizures may be present in the early neonatal period and may precede the onset of infantile spasms. In the present study, infantile spasms were associated with hypsarrhythmia in all 3 patients. Curatolo et al8 described awake, inter-ictal EEG showing focal or independent multifocal spike and slow wave activity at onset and later a pseudo-hypsarrhythmic pattern. They postulate that the infantile spasms in TSC may be a result of rapid secondary generalization. The presence of infantile spasms due to TSC is strongly predicted by the cortical tuber count, with earlier expression of parieto-occipital as compared to frontal cortical tubers.[8]
Hancock and Osborne[9] reviewed the efficacy and safety of vigabatrin in the treatment of infantile spasms. Of the 313 patients without TSC, 170 (54%) had complete cessation of their infantile spasms; of the 77 patients with TSC, 73 (95%) had complete cessation of their seizures. They concluded that vigabatrin should be considered as first line monotherapy for the treatment of infantile spasms in infants with either a confirmed diagnosis of TS or those at high risk i.e. those with a first-degree relative with TSC. Vigabatrin may paradoxically be less efficacious in those without TSC. Elterman et al[10] reconfirmed the efficacy and safety of vigabatrin in infantile spasms secondary to TSC, in a two-week randomized single-masked multicenter study with a three-year, open label dose ranging follow-up. Vigabatrin elevates GABA levels via irreversible inhibition of GABA transaminase. Its efficacy suggests that epileptogenesis in TSC may be related to an impairment of GABAergic transmission.
While it is effective in the treatment of infantile spasms, the use of vigabatrin is associated with a late appearance of visual field defects in upto 50% of patients. Currently, the minimum duration and doses of vigabatrin treatment that can produce side-effects are unknown. The feasibility of short treatment periods (2-3 months) is to be investigated.[11] In the present study, a two-and-a-half-year-old boy had delayed milestones and experienced recurrent myoclonic jerks which were poorly controlled with ACTH and valproate. There was significant improvement after adding vigabatrin and at follow-up two-and-a-half years later, he was free from seizures with a normal ophthalmic evaluation. Response to other anticonvulsants, however, is generally not very satisfactory.
Guerreiro et al[12] reviewed the results of surgery in 18 patients with TSC and medically refractory epilepsy. Twelve patients had well-localized epileptogenic lesions. The best outcome was obtained in patients who had local seizures and good imaging and EEG correlation. None of the patients in the present study was subjected to surgery.
Three types of nodular lesions, cortical tubers, subcortical heterotopic nodules and sub-ependymal giant cell astrocytomas have been described in the cerebrum of patients with TS.[13] Rao et al[1] in a detailed clinicopathological analysis of 6 cases of TS described cortical and sub-ependymal tubers, sub-ependymal giant cell astrocytoma, spongioblastoma, gemistocytoma and malignant vaso-formative tumor. MRI studies provide excellent in-vivo demonstration of the pathological lesions although CT scan is much less costly. Presently, MRI with gadolinium is recommended once every 2 years to follow growth of sub-ependymal nodules (SEN) smaller than 1 cm and for SENs greater than 1 cm, yearly MRI can best follow growth and development of ventricular enlargement. Conclusion Tuberous sclerosis is a diagnostic and therapeutic challenge. The revised diagnostic criteria enable more definite diagnosis. Hypsarrythmia in patients with TSC may be multifocal at onset. Seizures in TSC respond better to vigabatrin, however periodic evaluation of visual fields may be necessary. Patients with sub-ependymal nodules would require monitoring for early detection of obstructive hydrocephalus. Selected patients with focal seizures and good EEG-MRI correlation may benefit by surgical resection. The localization of TSC1 and TSC2 genes has further enhanced our understanding of this disease. It is likely that clinical research in the next few decades will further improve management. Acknowledgements The authors gratefully acknowledge the whole-hearted cooperation and support of Mr. M.V.Srinivasan, Mr. K.Bhaskar and Mr. N.Siddaraju, which made this work possible.
References
| 1. | Rao VT, Hegde T, Das S, et al. A clinicopathological study of Tuberous Sclerosis. Neurology India 1986;34:249-60. Back to cited text no. 1 |
| 2. | Verma BS, Tailor MH. Familial tuberous sclerosis: A review with report of three cases. Indian Paediatrics 1965;2:401-10. Back to cited text no. 2 [PUBMED] |
| 3. | Crino PB, Henske EP. New developments in the neurobiology of the tuberous sclerosis complex. Neurology 1999;53:1384-90. Back to cited text no. 3 [PUBMED] |
| 4. | van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSCI on chromosome 9q34. Science 1997;277:805-8. Back to cited text no. 4 [PUBMED] [FULLTEXT] |
| 5. | The European Tuberous Sclerosis Consortium: Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993;75:1305-15. Back to cited text no. 5 |
| 6. | MacCollin M, Kwiatkowski D. Molecular genetic aspects of the phakomatoses: Tuberous sclerosis complex and neurofibromatosis I. Current Opinion in Neurology 2001;14:163-9. Back to cited text no. 6 [PUBMED] [FULLTEXT] |
| 7. | Roach ES. Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: Revised clinical diagnostic criteria. J Child Neurol, 1998;13:624-8. Back to cited text no. 7 |
| 8. | Curatolo P, Seri S, Verdecehia M, et al. Infantile spasms in tuberous sclerosis complex. Brain Dev 2001;23:502-7. Back to cited text no. 8 |
| 9. | Hancock E, Osborne JP. Vigabatrin in the treatment of infantile spasms in tuberous sclerosis: Literature review. J child Neurol 1999;14:71-4. Back to cited text no. 9 [PUBMED] |
| 10. | Elterman RD, Shields WD, Mansfield KA, et al. US. Infantile spasms vigabatrin study group. Randomised trial of vigabatrin in patients with infantile spasms. Neurology 2001;23:57:1416-21. Back to cited text no. 10 |
| 11. | Curatolo P, Verdecchia M, Bombardieri R. Vigabatrin for tuberous sclerosis complex. Brain Dev 2001;23:649-53. Back to cited text no. 11 [PUBMED] [FULLTEXT] |
| 12. | Guerreiro MM, Andermann F, Andermann E, et al. Surgical treatment of epilepsy in tuberous sclerosis: Strategies and results in 18 patients. Neurology 1998;51:1263-9. Back to cited text no. 12 [PUBMED] |
| 13. | Mizuguchi M, Takashima S. Neuropathology of tuberous sclerosis. Brain Dev 2001;23:508-15. Back to cited text no. 13 [PUBMED] [FULLTEXT] |
Copyright 2004 - Neurology India
The following images related to this document are available:
Photo images
[ni04064f1.jpg]
|

{kind=link}