
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 52, Num. 2, 2004, pp. 254-256
|
Neurology India, Vol. 52, No. 2, April-June, 2004, pp. 254-256
Case Report
Congenital fiber type disproportion: A rare type of congenital myopathy: A report of four cases
Sharma MC, Ralte AM, Atri SK, Gulati S, Kalra V, Sarkar Chitra
Departments of Pathology, All India Institute of Medical Sciences, New Delhi
Correspondence Address:Professor of Pathology, Department of Pathology, All India Institute of Medical Sciences, New Delhi - 110029
sarkarcs@hotmail.com
Code Number: ni04077
Abstract Congenital fiber type disproportion is a rare type of congenital myopathy which presents as hypotonia, delayed motor milestones and dysmorphic facies. During the past 2 years we received 449 muscle biopsies, of which 4 cases were diagnosed as congenital fiber type disproportion (CFTD). In addition to CFTD, one case also had centronuclear features. Three of them were females and one was a male child. Although rare, it should be considered in the differential diagnosis of childhood muscle diseases. Histochemical staining is necessary for the diagnosis of this entity.
Keywords: Congenital myopathy, myopathic disorder, muscle,
congenital fiber type disproportion, enzyme histochemistry
Introduction Congenital fiber type disproportion (CFTD) is a rare type of congenital myopathy which was first described by Brooke[1] in 1973 and since then about 36 cases have been described in the literature.[2] This disease is transmitted as an autosomal recessive trait[3] but exceptions have been reported.[4],[5] CFTD is clinically characterized by hypotonia and delayed motor milestones, and histologically, Type I fibers are smaller than Type II fibers and there is a predominance of the former.
We describe four rare cases of CFTD which we encountered in the past two years. Case Report
Case 1
A 17-year-old female presented with history of progressive muscle weakness of both lower limbs which started at the age of 10 years. There was history of delayed motor milestones. Examination revealed normal facies, and muscle bulk, tone and power was normal. Electrophysiological studies (EPS) revealed myopathic pattern and normal creatine phosphokinase (CPK) level. Muscle biopsy was done with a clinical diagnosis of limb girdle muscular dystrophy.Case 2
A three and half-year-old-male child presented with history of inability to walk and feeding difficulties. He was the first child of a non-consanguineous marriage. Examination revealed long facies, eyelid drooping (ptosis), high arched palate and squint. Muscle tone was decreased and reflexes were absent. Genetic studies revealed no SMA gene deletions. CPK level was normal (CPK-59 IU/L). Muscle biopsy was done with a clinical diagnosis of congenital myopathy. Case 3
A 4-year-old female child presented with history of frequent falls and ptosis of 3 years duration. Antenatal history was uneventful and she was a product of a non-consanguineous marriage. Examination showed high arched palate, flat foot, ptosis and squint. Muscle tone was normal and EMG showed myopathic changes. CPK level was 182 IU/L. Muscle biopsy was done with a clinical diagnosis of congenital myopathy. Case 4
A 12-year-old female child presented with history of decreased movements since birth and delayed motor milestones. Examination revealed large head, low set ears, and facial dysmorphism. There was generalized hypotonia and reflexes were sluggish. EPS revealed myopathic changes and CPK was raised (455 IU/L). Pathological examination
Muscle biopsy in all cases shows similar features with minor variations. The majority of the fibers were small in diameter [Figure - 1]a but no angulated fibers were seen. A small percentage of fibers were either of normal diameter or appeared to be hypertrophied. There was no myophagocytosis and regeneration and degenerating fibers were not seen. Endomysial and perimysial fibrosis was minimal or absent.
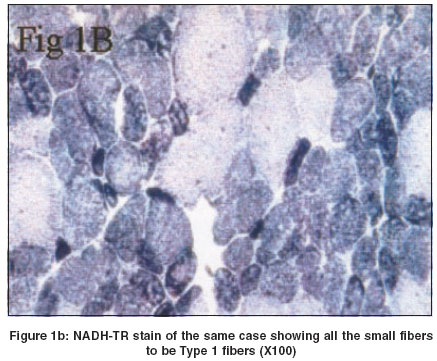
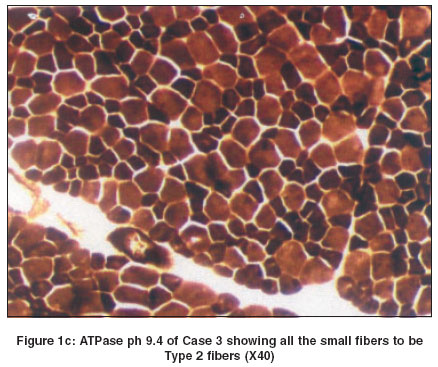
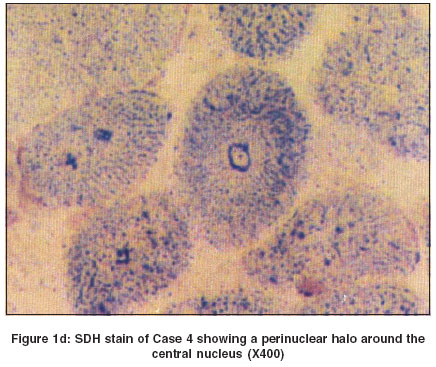
In all cases except Case 2, there was predominance of Type I fibers. This feature was better appreciated on NADH-TR [Figure - 1]b and ATPase stains. In Case 2 there was predominance of Type II fibers [Figure - 1]c. All the small fibers were of similar type. In addition to fiber type disproportion, Case 4 also revealed central nuclei in the majority of the fibers and oxidative stains revealed a perinuclear halo in this case [Figure - 1]d. Discussion Congenital fibre type disproportion (CFTD) is characterized by congenital hypotonia and delayed motor milestones, often associated with congenital dislocation of the hip joint, high arched palate, kyphoscoliosis and contractures. EMG shows myopathic pattern and the CPK level is normal or mildly elevated. Rarely, associated cardiomyopathies have been reported. Histologically, the muscle biopsy shows relatively smaller fibers (at least 25%) of Type I as compared to Type II fibers. Type II fibers are either of normal size or hypertrophied and there is a predominance of Type I fibres.[6],[7] All the cases under discussion, except Case 2 revealed hypoplasia and predominance of Type I fibers.
In addition, Case 4 showed central nuclei in a significant number of fibers. This has been reported earlier either at the time of first diagnosis[8] or in a repeat biopsy after an interval of 41 months.[9] Even a change from CFTD to myopathic features like LGMD has been described in the sequential biopsies.[2]
Interestingly, our Case 2 showed hypoplasia and predominance of Type II fibers and hypertrophy of Type 1 fibers.
Morphologically, a close differential diagnosis of this condition is neurogenic atrophy like spinal muscular atrophy (SMA). In the absence of atrophied and angulated fibers, hypoplasia of only Type I fibers and lack of type grouping the possibility of the latter is excluded.
We conclude that CFTD is a rare type of congenital myopathy and should be considered in the differential diagnoses of early onset myopathies. Histochemical staining is necessary for its diagnosis and differentiation from other myopathies and spinal muscular atrophy.
References
| 1. | Brooke MH. Congenital fibre type disproportion. Excerpta Medics 1973;295:147-59. Back to cited text no. 1 |
| 2. | Bartholomeus MG, Gabreels FJ, ter Laak HJ, van Engelen BG. Congenital fibre type disproportion a time-locked diagnosis: A clinical and morphological follow up study. Clin Neurol Neurosurg 2000;102:97-101. Back to cited text no. 2 [PUBMED] [FULLTEXT] |
| 3. | Cavanagh NPC, Larke BD, McMeniman P. Congenital fibre type disproportion myopathy. A histological diagnosis with an uncertain clinical outlook. Arch Dis Childhood 1979;54:735-43. Back to cited text no. 3 |
| 4. | Fardeau M. Congenital myopathies. In: Mastalgia FL, (Ed). Skeletal muscle pathology. New York: Churchill Livingstone 1992;237-8. Back to cited text no. 4 |
| 5. | Kinoshita M, Satoyoshi E. Type I fibre atrophy and nemaline bodies. Arch Neurol 1974;31:423-5. Back to cited text no. 5 [PUBMED] |
| 6. | Dubowitz V. Congenital fibre type disproportion - A pathology in search of a disease. Muscle disorders in childhood 1995;166-7. Back to cited text no. 6 |
| 7. | Schroeder JM. Congenital fibre disproportion. In: Lane RJM, (Ed). Handbook of muscle diseases. 1996:195-200. Back to cited text no. 7 |
| 8. | Gayathri N, Das S, Vasanth A, Devi MG, Ramamohan Y, Santosh Y, et al. Centronuclear myopahy - Morphological relation to developing clinicopathological evaluation. Neurol India 2000;48:19-28. Back to cited text no. 8 |
| 9. | Danon MJ, Giometti CS, Mamaligod JR, Swisher C. Sequential muscle biopsy changes in a case of congenital myopathy. Muscle Nerve 1997;20:561-9. Back to cited text no. 9 |
Copyright 2004 - Neurology India
The following images related to this document are available:
Photo images
[ni04077f1a.jpg]
[ni04077f1d.jpg]
[ni04077f1c.jpg]
[ni04077f1b.jpg]
|

![Figure - 1]](/showimage?ni/photo/ni04077f1a.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}