 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
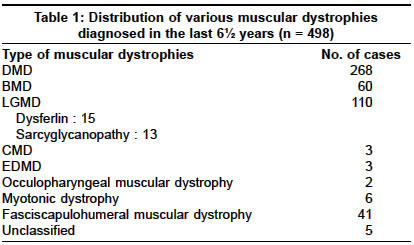
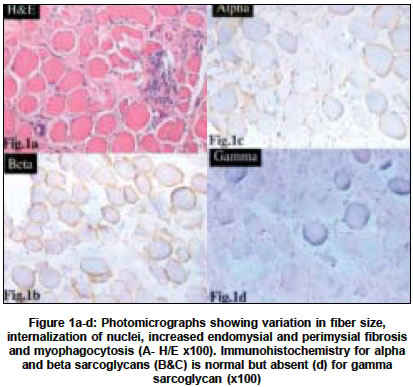
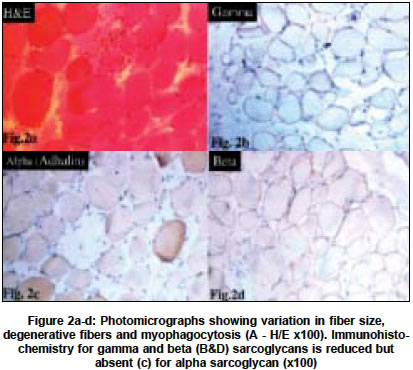
Neurology India, Vol. 52, No. 4, October-December, 2004, pp. 446-449 Original Article Sarcoglycanopathies: An enigmatic form of muscular dystrophy - A report of 7 cases Sharma MC, Mannan R, Singh NG, Gulati S, Kalra V, Sarkar Chitra Departments of Pediatrics, All India Institute of Medical Sciences, New Delhi - 110029 Code Number: ni04152 ABSTRACT BACKGROUND: Limb girdle muscular dystrophy (LGMD) is a phenotypic expression of a heterogeneous group of diseases and sarcoglycanopathy is one of the causes of LGMD. There is only one study on sarcoglycanopathies in the Indian literature. No data is available from northern India.MATERIALS AND METHODS: All cases of muscular dystrophies, which were diagnosed in our laboratory in the last six years, were reviewed. Immunohistochemistry for various sarcoglycan proteins was done. Clinical features and pathological findings of the cases that were diagnosed as sarcoglycanopathies were reviewed. RESULTS: In the last 6 ½ years (1998-June 2004), we received 1435 muscle biopsies, of which 498 cases were of muscular dystrophies, and 13 cases were of sarcoglycanopathies (8 of gamma, 3 of alpha, 1 of both alpha and gamma, and 1 with absence of all four sarcoglycans). Sarcoglycanopathies comprised 2.6% of all muscular dystrophies, 11.8% of LGMD and 0.90% of all muscle diseases diagnosed in our laboratory. The mean age of onset was 7.2 years and the M:F ratio was 1.1:1. Most of them presented with difficulty in getting up, climbing stairs, calf hypertrophy and markedly raised CPK levels. Histological features were like dystrophinopathies. CONCLUSION: Sarcoglycanopathies are a relatively rare cause of LGMD and should be confirmed by immunohistochemistry as it will facilitate counseling and also prognostification. Although rare, in patients with muscle weakness, calves hypertrophy and raised CPK levels this possibility should be considered and needs to be differentiated from dystrophinopathies. Key Words: Muscular dystrophy, sarcoglycanopathy, SCARMD, LGMD, histochemistry, muscle immunohistochemistry. INTRODUCTION Limb girdle muscular dystrophy (LGMD) has long been a controversial entity.[1],[2] It encompasses a variety of muscular dystrophies presenting with limb girdle weakness. Hence, it is a phenotypic expression of a heterogeneous group of diseases. The common phenotype expressed in LGMD includes muscle weakness progressing to muscle wasting of the limbs. Proximal muscles are more severely affected than distal muscles. The heart and bulbar muscles are spared except in rare instances. After the discovery of the dystrophin gene and its protein, the dystrophin-associated glycoproteins (DAP) were purified and characterized. The sarcoglycans are a group of 4 distinct integral membrane proteins, each from a distinct gene, that associate in a tetrameric complex, known as sarcoglycan complex. The complex is directly associated with dystroglycan and indirectly with dystrophin.[3] There are four sarcoglycan genes and their corresponding proteins, namely a,b,g, and d. These are transmembrane glycoproteins and range in size from 35 to 50 KD. The a,b,c,d, saroglycan complex is muscle-specific, however, a fifth protein e-sarcoglycan replaces a-sarcoglycan in some non-striated muscle tissues. Mutations in any one of the genes except epsilon result in autosomal recessive limb girdle muscular dystrophy (LGMD 2C-2F) which is clinically and pathologically indistinguishable from primary dystrophinopathies.[4] The function of the sarcoglycan complex is not exactly known. However, they are thought to be involved in the stabilization of the membrane and signal transduction. There is scarcity of data on the distribution of neuromuscular diseases in populations other than those in North America, Europe, Japan and Australia.[5] There is only one study in the Indian literature regarding deficiency of sarcoglycan proteins.[6] Hence, we undertook this study to describe the clinicopathological features of this entity. MATERIALS AND METHODS The muscle biopsies received in the Department of Pathology of this hospital between January 1998 to June 2004 (both retrospective as well as prospective) were reviewed and cases diagnosed as sarcoglycanopathies were included in this study. The clinical data was retrieved from the hospital files and muscle biopsies were reviewed. All muscle biopsies had been received in a fresh state without any fixative and divided into three portions. One portion was snap frozen in liquid nitrogen after putting in isopentane. Six-micron thick sections were cut for routine H/E staining, enzyme histochemistry (NADH-TR, SDH, MGT, COX with SDH and ATPase) and immunohistochemistry. The following antibodies were used: dystrophin 1, 2, 3 (dil 1:5), sarcoglycans a,b,g,d (dil 1:50), emerin (dil 1:25), Lamin A/C (dil 1:25), dysferlin (Hamlet,2], dil 1:25) and merosin (dil 1:50). All antibodies were obtained from M/s Novacastra, UK. The second portion of the muscle biopsy was routinely processed and paraffin embedded. Sections were stained with hematoxylin and eosin (H/E). Special stain, masson trichrome (MT) was done to demonstrate degree of endomysial and perimysial fibrosis. The third portion was fixed in 2.5% glutaraldehyde and post-fixed in osmium tetraoxide for electron microscopy. Ultrathin sections were cut and electron microscopy was done whenever required. In the cases which were negative for any of the sarcoglycans, IHC was repeated to confirm the diagnosis. Only cases which showed absence of staining for sarcoglycans were included in this study, in order to avoid individual subjectivity in staining interpretation in the absence of genetic studies. RESULTS During a period of 6½ years (January 1998-June 2004), 1435 muscle biopsies were received in our laboratory, out of which 498 (34.7%) cases were diagnosed as muscular dystrophies. The distribution of various muscular dystrophies is shown in [Table - 1]. The majority of the patients were of dystrophinopathies, limb girdle muscular dystrophies (LGMD), fascioscapulohumeral and others. Of 110 cases of LGMD, 15 were of dysferlinopathy and 13 of sarcoglycanopathies. Thirteen cases were diagnosed as sarcoglycanopathies, which constituted 2.4% of muscular dystrophies, 0.93% of all muscle diseases and 11.8% of LGMD diagnosed in our laboratory. The clinical data of the cases of sarcoglycanopathies is summarized in [Table - 2]. Age ranged from 5 to 20 years and the mean age at onset of symptoms was 7.2 years. All patients except one presented in the first decade of life. The male to female ratio was 1.1:1. All of them except one presented with difficulty in getting up, standing and climbing stairs. In all patients except one there was non-selective involvement of proximal muscles. Calves hypertrophy was observed in 9 patients (69.2%) and positive Gower′s sign in 7 patients (53.8%). None of our patients showed distal muscle involvement. CPK levels were markedly elevated in all of them. EMG revealed myopathic pattern in all of them and nerve conduction velocity (NCV) studies were normal. Pathologic examination All muscle biopsies showed loss of architecture, variation in fiber size of variable degree, central location of nuclei, split fibers, focal muscle fiber degeneration and myophagocytosis. There was endomysial and perimysial fibrosis with adipose tissue infiltration. Fibrosis was more prominent with Masson trichrome stain (MT). Degenerating fibers were non-reactive with NADH-TR and SDH oxidative stains. On NADH-TR staining lobulated fibers were seen in 3 cases (23%). Immunohistochemistry for sarcoglycans revealed negative staining for gamma in 8 cases, a- in 3 cases [Figure - 1] and [Figure - 2] and 1 case each of a, c and abcd. In 4 cases there was associated decrease in staining for other sarcoglycans (Cases 5,9],12],13). Staining for d sarcoglycans, Dys 1,2],3], emerin, dysferlin and merosin was normal in all cases except 2 cases which showed reduction in staining for dystrophin protein. In addition, one case showed complete absence of staining for all sarcoglycans whereas another was negative for a and g. DISCUSSION The successful identification of the dystrophin gene and its protein led to the discovery of the dystrophin-glycoprotein (DAG) complex and sarcoglycans are an important component of this complex. Analysis of muscle biopsy sample using specific antibodies to the various sarcoglycans has become the primary diagnostic tool for investigating this group of diseases. Antibodies against all sarcoglycans are commercially available and immunostaining can be done on frozen sections. In some cases, immunostaining will suggest which sarcoglycan is primarily involved, e.g. labeling with one antibody is absent and labeling with other is normal or reduced. However, many authors have shown that mutations of one sarcoglycan gene can lead to the deficiency of all components of this complex.[7],[8] This is due to the fact that mutations of a single sarcoglycan gene produces destabilization of the entire complex and secondary reduction of other sarcoglycan proteins. Thus, a genetic defect of any component of the protein complex should be detectable with an antibody against any of the sarcoglycan proteins.[9] But this is not necessarily a universal finding. It has also been observed that in patients with dystrophin gene abnormalities secondary reduction or deficiency of sarcoglycan occurs[9],[10],[11] and vice versa is also true. Two of the female patients in the present series who had c sarcoglycan deficiency also showed decreased staining for dystrophin protein-like BMD phenotypes. Therefore, immunostaining for sarcoglycans should be coupled with dystrophin protein. Some authors have suggested that in all patients of sarcoglycan-related muscular dystrophies, there is a deficiency of a-sarcoglycan in addition to individual sarcoglycan deficiency.[10] Therefore, preliminary staining for a-sarcoglycan is recommended to diagnose any sarcoglycan deficiency, however, some authors refuted this. This observation did not hold true in this series as an associated decrease in a-sarcoglycan staining was observed in three cases only. In the present study, the almost equal male to female ratio confirms the autosomal recessive mode of inheritance. The early age of onset of symptoms, repeated falls, difficulty in standing and getting up from a squatting position are some of the features which closely mimic dystrophinopathies. However, severe Duchenne type phenotype was seen in 2 patients and one case was reported in detail.[12] The calf muscle hypertrophy, positive Gower′s sign and markedly raised CPK levels were some of the misleading pointers towards the diagnosis of dystrophinopathies. Two children with Duchenne type phenotype were 5 and 6 years old and presented with difficulty in getting up and climbing stairs at 3 years of age. There was calf muscle hypertrophy, positive Gower′s sign and markedly raised CPK levels. One of the elder brothers was also suffering from a similar illness (Case 8. Immunohistochemistry revealed isolated a-sarcoglycan deficiency in one case and in another case all four sarcoglycans were absent but dystrophin proteins were normal. The child with a-sarcoglycan deficiency was diagnosed as severe childhood autosomal recessive muscular dystrophy (SCARMD). This type of sarcoglycanopathy has been described in communities with consanguineous marriages and has been reported from all over the world.[13],[14] The low prevalence of this entity in north India may probably be attributed to a low rate of consanguineous marriages. In the last 6 ½ years we encountered 13 cases of sarcoglycanopathies, which constituted 11.8% of all LGMD. The incidence varied from country to country. Hiyashi et al from Japan reported almost similar incidence and in their series 8.8% of LGMD were sarcoglycanopathies.[15] The reported incidence is as high as 25% in the Netherlands[16] and 55% in Brazil.[17] A study from Mumbai (western part of India) also reported a high incidence of sarcoglycanopathies. Khadilkar et al[6] reported 25 cases (46.2%) of sarcoglycanopathies in a series of 54 cases of LGMD occurring over a period of 5 years. The mean age of presentation was 25.8 years in their series and was high as compared to 7.2 years in the present series. In the present series, isolated g and a-sarcoglycan deficiency was seen in 8 and 3 patients respectively, while absence of both a, g and all sarcoglycans in one patient each. We believe that g and a sarcoglycanopathies are the most common types in India. Khadilkar et al described deficiency of multiple sarcoglycans in 84% of patients, while isolated deficiency of b- and d-sarcoglycans was noted in 12% and 4% patients respectively. However, none of our patients showed isolated deficiency of either b or d. The high frequency of sarcoglycanopathies in the series of Khadilkar et al can be attributed partly to selection bias as only adult patients were included in their series or it may be reflecting the truly high incidence from the State of Maharashtra, similar to some of the hemoglobulinopathies and hypo/afibrinogenaemia, which are reported more frequently in certain areas.[18],[19] The selective low incidence of sarcoglycanopathies from North India in the present series may not be absolutely true since we included only those cases which showed complete absence of staining for any of the sarcoglycans in order to avoid individual subjectivity in the interpretation of staining, in the absence of non-availability of genetic studies for sarcoglycans in India. Secondary reduction along with primary deficiency of sarcoglycans was seen in 4 cases. We observed that primary deficiency of one sarcoglycan could lead to reduction in the staining intensity of others but not with the complete absence of all other sarcoglycans, therefore, primary deficiency can be pinpointed by immunohistochemical staining and should be confirmed by genetic studies. Since no treatment is available presently, the future promising therapeutic modality is gene therapy. CONCLUSIONS Our study demonstrated the importance of immunohistochemistry in the diagnosis of sarcoglycanopathies. In patients, especially females, who present with muscle weakness, calf hypertrophy and raised CPK levels, the possibility of sarcoglycanopathy should be considered if staining for dystrophin is normal or reduced. ACKNOWLEDGEMENT The authors wish to thank Mr. Rajeshwar Khadia, Mr. Pankaj Kumar for their technical help and Mr. Kamal for secretarial assistance. REFERENCES
Copyright 2004 - Neurology India The following images related to this document are available:Photo images[ni04152t1.jpg] [ni04152f1.jpg] [ni04152f2.jpg] [ni04152t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}