 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
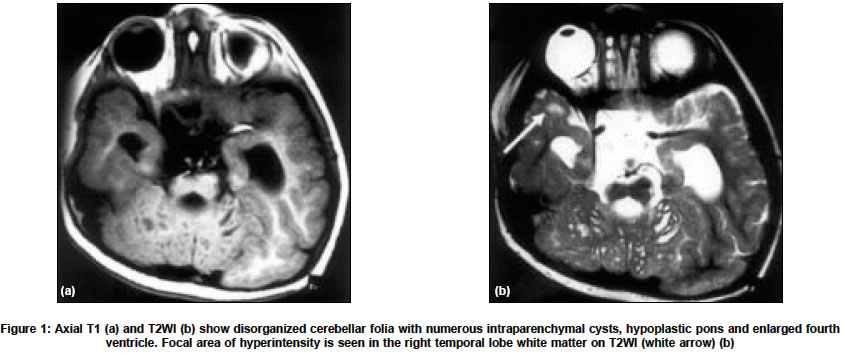
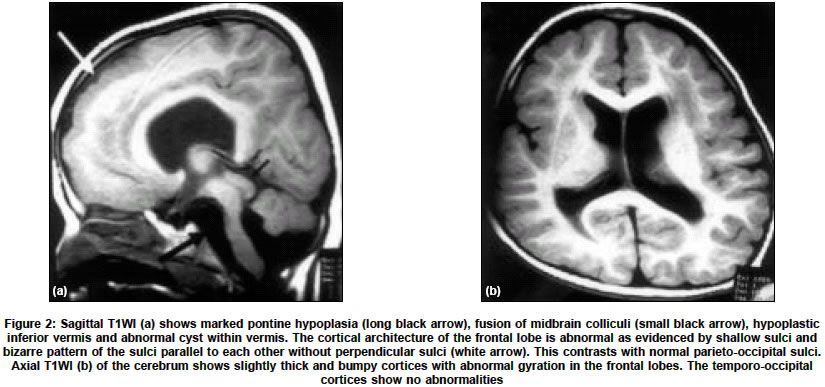
Neurology India, Vol. 52, No. 4, October-December, 2004, pp. 496-498 Case Report Congenital muscular dystrophy with characteristic radiological findings similar to those with Fukuyama congenital muscular dystrophy Garg Ajay, Gulati Sheffali, Gupta Vipul, Kalra Veena Departments of Pediatrics, AIIMS, New Delhi Code Number: ni04166 ABSTRACT Fukuyama congenital muscular dystrophy (FCMD) is the most common congenital muscular dystrophy in Japan and there are isolated reports of non-Japanese patients with FCMD. We report an Indian patient with congenital muscular dystrophy and characteristic radiological findings similar to those with FCMD.Key Words: Fukuyama congenital muscular dystrophy, congenital muscular dystrophy INTRODUCTION Congenital muscular dystrophy (CMD) is a heterogeneous group of disorders characterized by muscular hypotonia of prenatal onset and the histologic features of muscular dystrophy.[1] Based on the clinical, immunohistochemical, and genetic findings, eight different forms have been characterized genetically, and additional forms have been identified clinically.[2] Muscle-eye-brain disease (MEB), Walker-Warburg syndrome (WWS), and Fukuyama congenital muscular dystrophy (FCMD) are clinically similar autosomal recessive disorders characterized by congenital muscular dystrophy, lissencephaly, and eye anomalies.[3] FCMD is the most common congenital muscular dystrophy in Japan. Isolated cases of FCMD in non-Japaneses have been reported in other countries.[4],[5],[6],[7] We report an Indian patient of congenital muscular dystrophy with characteristic radiological findings similar to those with FCMD. CASE REPORT A three-and-a-half-year-old boy, born out of a non-consanguineous marriage, presented with hypotonia and developmental delay since birth. He was unable to stand, speak, and was incontinent. He acquired social smile at 2 months, partial head control at 8 months, started sitting with support at 9 months and sitting without support at 18 months. He had no seizures. On examination, he had head lag and hypotonia. Power was 3/5 in all limbs. Deep tendon jerks were not elicitable. There was no muscle hypertrophy. The ophthalmic examination, visual evoked potentials, motor nerve conduction studies and EEG were normal. Electromyography showed a myopathic pattern. Serum creatine kinase was elevated at 2970 U/l (normal 24-190 U/l). Cranial MRI revealed disorganized cerebellar folia with multiple cysts within vermis and cerebellar hemispheres [Figure - 1] a and b. There was marked pontine, and inferior vermian hypoplasia and fused midbrain colliculi [Figure - 2a]). The frontal lobes had shallow sulci and a bizarre pattern of the sulci parallel to each other without perpendicular sulci [Figure - 2a and b] . There was ventriculomegaly and abnormal increased signal in periventricular white matter. The corpus callosum had a bizarre configuration, though was not hypoplastic. In view of cortical dysplasia with characteristic distribution, cerebellar abnormalities, preserved general configuration of the brain and normal ophthalmologic examination, a diagnosis of FCMD was considered. Muscle biopsy and genetic analysis could not be done. The patient succumbed to respiratory infection and autopsy was denied. DISCUSSION Mutations of glycosylation enzymes have been commonly suggested in congenital muscular dystrophies (CMDs) with brain malformation and ocular involvement. These include the Muscle-eye-brain disease (MEB), Walker-Warburg syndrome (WWS), and Fukuyama congenital muscular dystrophy (FCMD) and the CMD due to fukutin-related protein (FKRP) defects.[3] The causative genes for these disorders are assumed to play a role in the glycosylation of dystroglycan, whose disruption can result in a decreased integrity of the dystrophin-glycoprotein complex and can lead to the dystrophic pathology of skeletal muscles and brain malformation.[3] The severity of the clinical and radiological findings of the present patient is most similar to that in typical FCMD cases. Patients with FCMD manifest weakness of the facial and limb muscles, and general hypotonia that usually appears before nine months of age.[1],[8] Most patients are never able to walk. Seizures occur in nearly half of the cases.[1] Severe metal retardation is observed in all cases. The patients survive beyond infancy, and ocular manifestations are rare and usually mild. Patients usually become bedridden before 10 years of age due to generalized muscle atrophy and joint contracture, and most of them die by 20 years of age.[1] These features are largely milder than the clinical course of patients with WWS and MEB, but are more severe than those with FKRP defects. On the other hand, what is confusing is the overlap between the genotype-phenotype correlations of the disorders. A homozygous nonsense mutation in the Fukutin gene has been reported in a Turkish patient with WWS phenotype.[9] Similarly, mutations in the FKRP give rise to a broad clinical spectrum that ranges from mild muscular dystrophy (LGMD2I) to the more severe form (MDC1C), which in some cases is also associated with intellectual impairment and cerebellar cysts.[10] Thus the term FCMD should be used to describe the clinical phenotype, and not the genotype of individual patients. We diagnosed the present patient as having FCMD based on these considerations, on the clinical severity, including the findings on MR images as discussed below. The most common and characteristic changes in the central nervous system are brain malformations, which include polymicrogyria, pachygyria, and agyria of the cerebrum and cerebellum (cobblestone cortex) lacking neuronal lamination of the normal six-layered cortex.[3],[4],[11] In brain MR examination, all patients show polymicrogyria, and approximately half the patients show pachygyria. These two types of cortical dysplasias are present in characteristic distributions: the former demonstrates frontal lobe involvement in all and parietotemporal lobe involvement in some, whereas the latter involves the temporo-occipital lobes.[4] Most patients show prolonged T1 and T2 signal in the white matter, which is thought to be due to delayed myelination.[3] Various degrees of ventriculomegaly and hypoplasia of the pons can be seen sometimes.[3],[4] Cerebellar polymicrogyria depicted as disorganized cerebellar foliation accompanying cysts is found in more than 90% of the patients.[4] Both lesions tend to be located in the mid portion and dorsal surface of the hemisphere, particularly in the superior semilunar lobule. WWS and MEB can also cause similar MR findings. However, FCMD is associated with less severe gyral malformations and cerebellar anomalies than WWS, and ocular involvement is less severe in FCMD.[11] Patients with WWS show diffuse cerebral cobblestone cortex, absence of cerebral and cerebellar myelin, cerebellar polymicrogyria (with or without cysts), hydrocephalus, pontine and cerebellar vermian hypoplasia.[11] The septum pellucidum is absent, and the corpus callosum, basal ganglia and thalami are markedly hypoplastic. Patients with MEB show cerebellar polymicrogyria (with or without cysts), absence of the septum pellucidum, diffuse cerebral cortical dysplasia, pontine and cerebellar vermian hypoplasia, patchy hypomyelination, and variable callosal hypogenesis and hydrocephalus. Traditionally, the diagnosis is based on pathologic evidence of muscular dystrophy in the biopsy specimen obtained in the appropriate clinical context.[4] Nevertheless, the presence of brain malformation can be overlooked in the presence of muscular symptoms, and a muscle biopsy does not provide specific evidence for FCMD beyond the mere diagnosis of muscular dystrophy.[4] The diagnosis of FCMD can be facilitated with MRI, which readily demonstrates a variety of brain malformations in FCMD, the constellation of which is considered to be diagnostic of this entity without a muscle biopsy.[4] In summary, the index patient presented with muscular hypotonia and developmental delay. He had normal ophthalmologic examination. MR imaging showed disorganized cerebellar folia with cysts, marked pontine hypoplasia, bilateral frontal cortical dysgenesis, ventriculomegaly, fused midbrain colliculi and white matter T2-hyperintensity. In view of cortical dysplasia with characteristic distribution, cerebellar abnormalities, preserved general configuration of the brain and normal ophthalmologic examination, a diagnosis of FCMD was made. We speculate that this disorder may be more common than is generally recognized and that the use of MR imaging and increased awareness among radiologists will help to identify these patients more readily. REFERENCES
Copyright 2004 - Neurology India The following images related to this document are available:Photo images[ni04166f2.jpg] [ni04166f1.jpg] |
| |||||||||

{kind=link}
{kind=link}