
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 53, Num. 1, 2005, pp. 102-104
|
Neurology India, Vol. 53, No. 1, January-March, 2005, pp. 102-104
Case Report
Hallervorden spatz disease: MR and pathological findings of a rare case
Sharma MC, Aggarwal N, Bihari M, Goyal V, Gaikwed S, Vaishya S, Sarkar Chitra
Departments of Pathology, All India Institute of Medical Sciences, New Delhi
Correspondence Address:Department of Pathology, All India Institute of
Medical Sciences, Ansari Nagar, New Delhi - 110 029 Email: sarkarcs@hotmail.com
Code Number: ni05029
Abstract We describe a child with pathologically proven Hallervorden Spatz disease. He presented with extrapyramidal symptoms and characteristic "eye-of-the-tiger" sign on magnetic resonance imaging. He was given the possible benefit if any of deep brain stimulation with no much improvement. Pathological examination of the brain showed iron deposition in bilateral globus pallidi, spongiform change and neuron axonal degeneration (spheroids).
Key Words:
Hallervorden Spatz disease, movement disorder, pantothenate kinase 2 deficiency
Introduction
Hallervorden Spatz disease (HSD) is a rare neurodegenerative
disorder of basal ganglia and is characterized by extrapyramidal symptoms,
mental deterioration, dementia, and retinal degeneration. Both familial
and sporadic cases have been reported. Only six cases have been reported
from India, clinical diagnosis based on clinical and magnetic resonance
imaging characteristics.[1],[2],[3],[4] [Table
- 1]. We report a pathologically proven case of HSD from India.
Case Report
A 8-year-old boy presented with a 2 year history of abnormal flexor posturing
of the right hand and wrist with clenching of the fist. Over 6 months it
became fixed and persistent and also involved the left hand. Subsequently,
he developed extension of the neck and flexion of the trunk, with grimacing
of the face, tight closure of the mouth and deterioration of speech and
walking. He was the product of a non-consanguineous marriage and was born
at full term. The labor was prolonged. His developmental milestones were
delayed and speech was slurred with inability to speak difficult words.
He was mentally retarded with poor scholastic performance and was sent
to a school for mentally challenged children. No other family members had
similar or other movement disorders. With levodopa with carbidopa he had
a mild improvement. Later baclofen and trihexyphenadyl were added. He continued
to worsen progressively.
On examination, he was bedridden with generalized dystonia, fixed contractures of the lower limbs and was incontinent. There was no evidence of Kayser-Fleischer ring or retinitis pigmentosa. Serum ferritin, ceruloplasmin levels were normal and blood smear was negative for acanthocytes. Brain commuter tomography and MRI (low field strength) done 2 years back were normal. Repeat MRI showed hypointensity with an area of central hyperintensity ("eye-of-the-tiger"-sign)
in both globus pallidi on T2 weighted imaging [Figure
- 1]. Based on the clinical and MRI features a diagnosis of Hallervorden Spatz Disease was made.
In view of poor response to pharmaco-therapy he was given the possible benefit if any of deep brain stimulation (DBS). Electrodes were implanted in bilateral globus pallidus interna. Postoperatively he developed severe stridor and could not be extubated and needed a tracheostomy. He developed pneumonia and died three months after the procedure.
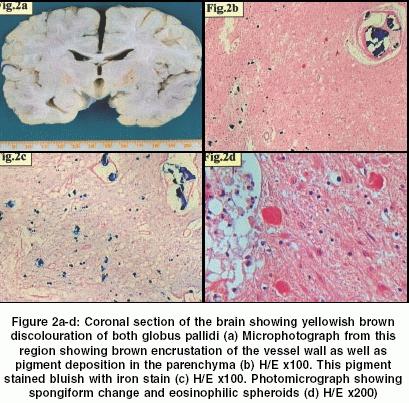
At autopsy the brain weighed 1100 grams before fixation with an unremarkable external surface. Coronal slicing of the brain showed brownish yellow discolouration of the bilateral globus pallidi. [Figure
- 2]a. Microscopic examination of the cerebellum and different
lobes of the cerebral hemispheres revealed slight prominence of melanin
containing cells in the leptomeninges without any hypoxic changes. Sections
examined from the globus pallidi showed brownish black color pigment deposition
in the parenchyma both extracellularly and intracellularly in the neurons
and astrocytes, and as mulberry-like encrustation of blood vessel wall
[Figure - 2]b. This pigment on Pearl′s
reaction proved to be iron [Figure
- 2]c and did not stain with the calcium stains. There was spongiosis, loss of neurons, gliosis and eosinophillic spheroids [Figure
- 2]d (neuron axonal dystrophy, NAD). These spheroids were immunoreactive to neurofilament. Iron deposition and spheroids were also seen in the corticostriate tracts but were not associated with demyelination or axonolysis of the tracts and white matter. Sections from the electrode site implantation revealed cyst formation and collection of foamy macrophages along with reactive gliosis of the surrounding parenchyma. These features were consistent with HSD.
Discussion
Hallervorden Spatz Disease (HSD) is a rare autosomal recessive neurodegenerative disorder with aberrant iron metabolism in the brain, first described by Hallervorden and Spatz in 1922. It is characterized by childhood onset of extrapyramidal motor symptoms. Some patients may present with mental changes, dementia and vision disturbances. Average survival after diagnosis onset is 11.8 years. Pathological findings include iron deposition, axonal swellings or spheroids (NAD) predominantly in the globus pallidus and pars reticularis of the substantia nigra. Since the first description of this disease, little progress has been made in the treatment.
Recently, the gene for the disease has been localized to chromosome 20p12.3-13, coding for pantothenate kinase 2[5] which is required for the phosphorylation of pantothenic acid in the formation of coenzyme A. Due to defective phosphorylation of pantothenic acid there is under utilization of cystine which, when in excess causes chelation of iron leading to free toxic radicals production. The preferential involvement of basal ganglia is attributed to the excess of pantothenate kinase receptors. Thus, the term pantothenate kinase 2-associated neurodegeneration (PKAN) may be preferable instead of HSD.[6]
The characteristic MR finding of "eye-of-the-tiger"-sign corresponds
to the pathological findings. The hypointensity on T2 weighted image
is because of iron deposition and central hyperintensity is secondary
to gliosis and spongiosis.[7] This
is well corroborated pathologically in this case also. The other conditions
in which high signal intensity, like HSD can be observed are metabolic
disorders, like organic acidurias, early onset levadopa responsive Parkinsonism
and cortical-basal ganglionic degeneration. The other disorder affecting
basal ganglia such as Leigh′s disease, mitochondrial encephalopathies, infantile bilateral necrosis and Wilson′s
disease more frequently involve the putamen rather than the globus pallidus.
The other differential diagnosis of iron deposition in the basal ganglia
and "eye-of-the-tiger"-sign include aceruloplasminemia and neuroferritinopathy.
These are distinct conditions of abnormal iron metabolism but unlike
HSD present in adult or late life. Neuroferritinopathy is characterized
by onset at 40-55 years of age and defect is localized to gene encoding
ferritin light chain polypeptide at 19q13.3. A ceruloplasminemia is associated
with diabetes mellitus and there is complete deficiency of ceruloplasmin
protein. The gene is localized to chromosome 3q13.3. Recently, neurodegenerative
diseases of brain with accumulation of iron have been classified according
to the age of onset and gene defect into different groups [Table
- 2]. Hayflick et al[8] studied
123 cases from 98 families and classified HSD clinically as classic disease
and atypical form. Classical HSD is characterized by early onset, rapid
progression and presence of typical "eye-of-the-tiger"-sign with PANK2 mutations. In contrast, atypical disease is characterized by late onset with slow progression and only one-third of the cases showed PANK2 mutations. "Eye-of-the-tiger"-sign may or may not be present. They concluded that all patients with "eye-of-the-tiger"-sign,
whether classic or atypical, showed PANK2 mutations and this favoured
to the diagnosis of HSD.
Management is symptomatic and there is no definitive treatment of this disease. Resistance, drugs adverse effects and ineffectiveness of the medical treatment in stopping the disease progression in movement disorders has led to exploration of surgical modalities in the treatment of these disorders. The role of surgical treatment for dystonia is evolving. Stereotactic pallidotomy[9] and thalamotomy[10] have been tried with good short-term results. However, these are permanent procedures with increased risk of side effects. In contrast, DBS is a relatively newly described technique, which is reversible and is seemingly free of side effects and complications apart from the risk of infection. It is however expensive. The principle of this technique is based on the concept that high frequency stimulation of neural cells lead to suppression or modulation of their activity, without generating irreversible anatomical lesions. Bilateral DBS was tried in this case but without any benefit. Future therapeutic strategies may involve direct delivery of phosphorylated pantothenate to the cells bypassing pantothenate kinase. Neuroprotection by the brain permeable iron chelator, VK-28 which inhibits both basal and Fe/ascorbate induced mitochondrial membrane lipid peroxidation, has shown promising results in rats.[11] Its potency is comparable to proteolytic iron chelator, desferal, which does not cross the blood-brain-barrier.
Thus, HSD is a rare neurodegenerative disorder characterized by iron
deposition in the globus pallidus with characteristic radiological "eye-of-the-tiger"-sign.
Pre- and post-natal molecular diagnosis is possible. The role of DBS
needs to be evaluated on large number of patients before it is discarded.
Acknowledgements
Authors are thankful to Dr. P. K. Ghosh, Associate Professor, Department
of Pediatrics, PostGraduate Institute of Medical Education and Research,
Chandigarh, India who discussed this case in Clinicopathological Conference
held on 29th October 2003 at All India Institute of Medical Sciences, New
Delhi, India.
References
| 1. | Kaushik A, Longia S, Jagadeesh R, Kishore V. Hallervorden-spatz disease. Indian Pediatr 1995;32:483-5. Back to cited text no. 1 |
| 2. | Singhi PD, Mitra S. Hallervorden spatz disease: Late infantile type. J Child Neurol 1997;12:281-2. Back to cited text no. 2 [PUBMED] |
| 3. | Shah J, Patkar D, Patankar T, Krishnan A, Prasad S, Limdi J. Hallervorden spatz disease: MR imaging. J Postgrad Med 1999;45:114-7. Back to cited text no. 3 [PUBMED] [FULLTEXT] |
| 4. | Rao C, Murthy V, Hedge R, Asha, Vishwanath. Hallervorden spatz disease. Indian J Pediatr 2003;70:513-4. Back to cited text no. 4 |
| 5. | Zhou B, Westway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel patothenate kinase gene (PANK2) is defective in Hallervorden spatz syndrome. Nature Genetics 2001;28:345-9. Back to cited text no. 5 |
| 6. | Hayflick SJ. Pantothenate kinase associated neurodegeneration (formerly Hallervorden-Spatz Syndrome). J Neurol Sci 2003;207:106-7. Back to cited text no. 6 [PUBMED] [FULLTEXT] |
| 7. | Adams RJ, Nichols FT, McKie V, McKie K, Milner P, Gammal TE. Hallervorden spatz syndrome: Clinical and magnetic resonance imaging correlations: Ann Neurol 1988;24:692-4. Back to cited text no. 7 |
| 8. | Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KH, et al. Genetic, clinical and radiographic delineation of Hallervorden-Spatz Syndrome. N Engl J Med 2003;348:33-40. Back to cited text no. 8 [PUBMED] [FULLTEXT] |
| 9. | Justesen CR, Penn RD, Kroin JS, Egel RT. Stereotactic pallidotomy in a child with Hallervorden- spatz disease. J Neurosurg 1999;90:551-4. Back to cited text no. 9 [PUBMED] |
| 10. | Tsukamoto H, Inui K, Taniike M, Nishimoto J, Midorikawa M, Yoshimine T, et al. A case of Hallervorden-spatz disease: progressive and intractable dystonia controlled by bilateral thalamotomy. Brain Dev 1992;14:269-72. Back to cited text no. 10 [PUBMED] |
| 11. | Shachar DB, Kahana N, Kampel V, Warshawsky A, Youdim MB. Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydrodopamine lesion in rats. Neuropharmacology 2004;46:254-63. Back to cited text no. 11 [PUBMED] [FULLTEXT] |
Copyright 2005 - Neurology India
The following images related to this document are available:
Photo images
[ni05029f2.jpg]
[ni05029f1.jpg]
[ni05029t2.jpg]
[ni05029t1.jpg]
|

![[Table - 1]](/showimage?ni/photo/ni05029t1.jpg){kind=link}
![[Figure - 1]](/showimage?ni/photo/ni05029f1.jpg){kind=link}
{kind=link}
![[Table - 2]](/showimage?ni/photo/ni05029t2.jpg){kind=link}