
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 53, Num. 2, 2005, pp. 156-161
|
Neurology India, Vol. 53, No. 2, April-June, 2005, pp. 156-161
Original Article
Neuropathological spectrum of Rasmussen encephalitis
Deb Prabal, Sharma MeharChand, Gaikwad Shailesh, Tripathi Manjari, Chandra PSharat, Jain Satish, Sarkar Chitra
Departments of Pathology, All India Institute of Medical
Sciences, New Delhi
Correspondence Address: Department of Pathology, All India Institute
of Medical Sciences, Ansari Nagar, New Delhi – 110 029, sarkarcs@hotmail.com
Date of Acceptance: 18-Apr-2005
Code Number: ni05045
Related article: ni05046.
ABSTRACT
Background : Rasmussen encephalitis (RE) is a chronic epileptic disorder of unknown etiology, and is clinically characterized by progressive neurological deterioration, focal seizures often progressing to intractable epilepsy, cognitive decline and hemispheric atrophy.
Aims : We report the spectrum of neuropathological changes seen in RE, and discuss the evolutionary mechanisms of this disease.
Materials and Methods: Surgically obtained specimens from RE patients diagnosed during 2002-2004 at this institute were evaluated for the presence and extent of different histopathological features of RE. The H&E and immunohistochemistry stained slides were also evaluated for the type and distribution pattern of inflammatory infiltrates, along with a semiquantitative evaluation for the severity of inflammation.
Results : Four cases of RE were admitted during the study period, all of which presented with partial seizures with successive deterioration to intractable epilepsy. The age at onset varied between 5 and 10 years (mean 7.8 years), with three male and one female patients. Subsequently, all four patients underwent hemispherotomy. Histopathological features of perivascular lymphocytic infiltrate, neuronal loss, microglial nodules, and reactive astrocytosis, with or without evidence of neuronophagia confirmed a diagnosis of RE. These cases also had varying degrees of T-cell-rich (CD3-positive) inflammatory infiltrates and CD68-immunopositive microglial proliferation. It was observed that the severity of inflammation had a trend to inversely correlate with the duration of symptoms.
Conclusion : It is proposed that an accurate evaluation and histopathological grading of these lesions may possibly have a role in patient prognostication.
Keywords: Encephalitis, intractable epilepsy, neuropathology, Rasmussen encephalitis
Rasmussen encephalitis (RE) is an unusual seizure disorder of unknown etiology first described by Rasmussen in 1958.[1] It is refractory to anticonvulsant therapy, and clinically characterized by progressive unilateral neurological deficit, with histomorphology of chronic encephalitis.[2]
Though RE may be suspected on the basis of clinical profile and radiological features, histopathology remains the gold standard for establishing the diagnosis.[3] Biopsy from a case of RE typically reveals a spectrum of inflammatory changes ranging from ′ active phase′ to ′ chronic phase ′ depending on the interval between onset/diagnosis and surgery.[2],[4]
Earlier, a series of five cases of progressive epilepsy due to chronic viral encephalitis with clinical profile resembling RE was reported from this institute.[5] However, to the best of our knowledge, we report the first Indian series of four cases of histopathologically proven RE, highlighting the neuropathological spectrum characteristic of such cases.
Though this is a cross-sectional study with tissue obtained at a single point of time in each patient, it gives an insight into the evolutionary mechanisms of the disease.
Materials and methods
Patients with the clinical course and neuroradiological features typical of RE treated at this institute during 2002-2004 were selected. Relevant clinical and radiological data in all cases were analyzed.
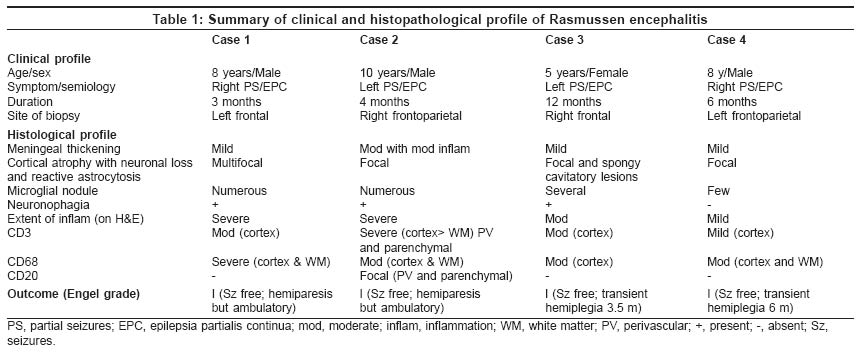
Specimens were collected during hemispherotomy (according to Delalande), from the cortical and subcortical areas of the affected lobes [Table
- 1].
Hematoxylin and Eosin (H&E) stained slides retrieved from the archives
were reviewed independently by three pathologists and diagnosis reconfirmed.
The number of blocks available per case ranged between 1 and 15 (mean
7). One representative block (displaying maximum inflammation and microglial
proliferation) of formalin-fixed paraffin-embedded tissue in each of
the four cases was selected; five-micrometer serial sections cut and
immunohistochemistry (IHC) performed by streptavidin-biotin immunoperoxidase
technique (LSAB Kit, M/sDakopatts, Denmark) using monoclonal antibodies
to CD3 - T-cell marker (1: 200, M/sDakopatts), CD20 - B-cell marker (1:
200, M/sNeomarker), and CD68 - macrophage marker (1: 100, M/sDakopatts).
Cases were classified into four broad groups, as suggested by Robitaille,[4] depending on the presence and extent of different morphological features diagnostic of RE, via, meningeal thickening, cortical atrophy with neuronal loss, and reactive astrocytosis, perivascular lymphocytic cuffing, microglial nodule formation and neuronophagia.
Group 1 : ′ Active disease ′showing perivascular
lymphocytic cells, numerous microglial nodules, with or without neuronophagia
and glial scarring.
Group 2 : ′ Active and remote disease ,′containing
several microglial nodules with perivascular round cell infiltrates,
with at least one gyral segment showing complete necrosis and cavitation
involving full thickness of the cortex.
Group 3 : ′ Remote disease ,′manifesting
as neuronal loss with reactive gliosis, and moderately abundant perivascular
inflammatory cells with few microglial nodules.
Group 4 : Showing ′ nonspecific change ′like
mild perivascular inflammation and microglial nodules, associated with
neuronal loss and glial scarring of varying degree.
The H&E and IHC stained slides were evaluated for the type of inflammatory
infiltrates along with their pattern of distribution. A semiquantitative
evaluation was also done to grade the severity of inflammation, on a
scale of mild to severe inflammation.
RESULTS
Four cases of RE were diagnosed during the study period. All patients
presented with partial seizures with duration ranging between 3 and 12
months (mean 6 months), and subsequently deteriorating to epilepsia partialis
continua (EPC). The mean age of the patients was 7.8 years (range 5-10
years). There were three male and one female patients. [Table
- 1] summarizes the clinical profile of these patients.
Magnetic resonance imaging (T1WI and T2WI) revealed cortical atrophy
of the affected hemisphere in all four cases [Figure
- 1]A, B. Further, Case 4 also showed a hyperintense signal in the medial frontal, occipital, and parietal lobes on the left side in addition to atrophic changes in the frontal and temporal lobes [Figure
- 2]A, B.
Biopsies from all four cases showed features typical of RE.
Cases 1 and 2 : Both biopsies yielded two bits of tissue each, which in total measured 3 x 2 x 1 cm and 6 x 4 x 2 cm, respectively.
Both biopsies showed cortical atrophy due to neuronal loss and astrocytic
proliferation. Evidence of ongoing inflammatory activity was characterized
by marked perivascular [Figure
- 3] and parenchymal CD3-immunopositive lymphocytic aggregates, mostly
confined to the cortex [Figure
- 4]A, B. However, Case 2 showed infiltration into the white matter,
along with focal CD20-immunopositivity [Figure
- 5].
This was complemented by moderate to severe degree of CD68-immunopositive
microglial proliferation forming microglial nodules, along with neuronophagia.
The above features were consistent with ′ active ′RE.[6]
Case 3 : Biopsy comprised two bits of tissue measuring 7 x 2.5 x 1 cm in total.
Sections revealed focal cortical atrophy associated with severe neuronal
loss, spongy changes, and cavitatory lesions in the cortex. There was
a moderate degree of perivascular T-lymphocytic infiltration and microglial
proliferation, along with the presence of several microglial nodules
and evidence of neuronophagia. These features were consistent with ′ active and remote ′RE
disease.[6]
Case 4 : Resected tissue measured 3 x 2.5 x 1 cm sections revealed
focal cortical atrophy with neuronal loss and gliosis. Inflammatory activity
was minimal, with mild T-cell infiltration in the superficial and deep
neuronal layers of the cerebral cortex located in the perivascular region.
Microglial activation was moderate with some microglial nodules but absence
of neuronophagia. The above features were consistent with ′ remote disease ′of
Robitaille.[6]
Correlation of disease duration with degree of inflammation
Cases 1 and 2 had shorter disease duration (3 and 4 months, respectively)
and showed severe degree of T-cell predominant inflammation, with microglial
proliferation forming numerous microglial nodules (′ active ′disease).
Similarly, Case 3, which showed a moderate degree of lymphocytic inflammation
and microglial proliferation, along with fewer microglial nodules, had
a relatively longer disease duration of 12 months (′ active and remote ′disease).
Though the severity of inflammation in the above cases inversely correlated
with disease duration, Case 4 was an exception, and displayed lesser
degree of inflammation, despite shorter disease duration than Case 3.
The follow-up period ranged between 8 and 14 months (mean 10 months).
In the immediate post-operative period, Cases 1 and 2 developed hemiparesis,
while Cases 3 and 4 had transient hemiplegia. Motor dysfunction was apparently
more marked in the distal upper limbs than in the proximal part, and
in the lower limbs. Muscle power in the distal upper limbs gradually
improved from an immediate postoperative level of 3/5 to 4 - 4 +/
5. None of the patients had clinical features of hemianopia, either historically
or on bedside testing, nor did they have persistent aphasia, during a
mean long-term follow-up period of 10 months. Currently, all cases are
seizure free and in Engel Grade I.
DISCUSSION
Diagnosis of RE is generally based on the characteristic clinical, radiological and pathological features.[2],[3] Typically, it affects previously healthy children and adolescents between the ages of 14 months and 14 years.[2] However, adults may rarely be affected.[4] It usually presents as pharmacoresistant generalized tonic-clonic or partial seizures, with about half of them subsequently progressing to EPC.[3],[4] Other manifestations include progressive unilateral neurologic deficit leading to hemiparesis and cognitive decline. Occasional cases of bilateral involvement have also been reported.[7] Dysphasia may develop if the dominant hemisphere is involved.[2] All the cases in the present series were in the pediatric age group, clinically characterized by AED-refractory partial seizures progressing to EPC.
Generally, RE cases have a temporal or frontoparietal onset, as judged by early MRI, which is concordant with the report that the most common partial seizures in RE are of the psychomotor and focal motor type.[3] A similar pattern of onset was also evident in all cases of the present series.
Clinically suspected RE cases routinely undergo MRI evaluation,[3],[8] since not all investigators consider brain biopsy essential for establishing a diagnosis. The characteristic MRI features in RE are areas of cortical hyperintense T2/fluid-attentuated inversion recovery (FLAIR) signal.[3],[9] Serial MRI studies[3],[10] have demonstrated a spectrum of changes ranging from an apparently normal image at onset, to focal or hemispheric cortical atrophy of the affected lobes, in the late stage. Neuroimaging in the present series showed evidence of cortical atrophy of the affected hemisphere, which suggested a fairly advanced disease process. Case 4, however, demonstrated more severe affection, which subsequently correlated with the advanced stage seen on histopathology.
Pathological appearances of RE, both macroscopic and microscopic, vary according to the severity of the disease. Macroscopic features in cases operated within 1-2 years of onset show subtle change in the form of slight discoloration and granularity, along with focal thinning of the cortex, while severe cases may show gyral atrophy with widespread hemiatrophy and ventricular dilatation.[6]
Despite a strong suspicion based on clinical and imaging features, histopathology has remained the gold standard for confirming a diagnosis of RE.[3] Classically, changes are restricted to the affected hemisphere, with sparing of the contralateral hemisphere, basal ganglia and posterior fossa.[2] However, head of the caudate nucleus is often affected by the atrophic process.[10] The
neuropathological spectrum of changes seen in the present series comprised ′ active ′disease
in Cases 1 and 2, characterized by severe inflammatory infiltrates/microglial
proliferation, while Cases 3 and 4 were designated as ′ active and remote ′and ′ remote ′disease,
respectively.
Robitaille[4] and Pardo et al[11] described the effect of disease duration on the burden of pathology. The highest inflammatory intensity and microglial proliferation with nodule formation are generally seen in the early stages, followed by decrease in the later stages. The intensity of inflammation, as represented by collection of T cells and microglial proliferation, has been reported to bear an inverse correlation with the disease duration.[11] Since the number of cases in the present study was very few, no definite statistical correlation was possible; however, the trend of inverse correlation of disease duration with severity of inflammation was evident in the first three cases, with Case 4 apparently being an exception.
Recently, Pardo et al .[11] reported the neuropathological profile in 45 RE cases, based on the disease stages for RE patients established by Robitaille.[4] They concluded that the age of onset of symptoms correlated with the burden of pathology and that the duration of disease process produced a significant change in the magnitude of pathologic changes, which was also noted in the present study.
Though the clinical profile of RE is not of an acute encephalitic illness, histopathology is strongly suggestive of a viral aetiology.[1] However, in situ hybridization and polymerase chain reaction studies have failed to identify any viral agent.[12] Some[13] have suggested an immune-complex deposition and vasculitis, or a coexisting second pathology,[14] as events leading to RE. Others[15] have demonstrated circulating autoantibodies against the GluR3 protein of glutamate receptor, thus favoring an autoimmune mechanism, although Watson et al .,[16] have failed to detect such antibodies in RE patients. Recently Bien et al .[17] have
hypothesized a cytotoxic CD3+CD8+ T-cell mechanism in the
pathogenesis of RE. It was interesting to note that all the cases in
the present study displayed CD3-immunopositive lymphocyte infiltrates,
while CD20 (B cells) were largely absent.
The unihemispheric pathology of RE, as reported in the literature (and also in the present study), has been explained due to a possible cytotoxic T-cell response, which is induced by a progressive disruption of the blood-brain barrier by an initial focal event, with subsequent spreading across the affected hemisphere.[17]
RE cases are typically AED-resistant, and have been managed surgically.[2] Procedures like functional hemispherectomy have been reported to be more effective in controlling seizures in patients with a largely destroyed hemisphere and residual hemiplegia,[18] in comparison to focal excision, partial corticectomy, and subtotal hemispherectomy. Patients in the present series were also resistant to AEDs and had been successfully managed by hemispherotomy according to Delalande.
Immunosuppressive and immunomodulatory treatments have been suggested to be most useful during stages of active inflammation,[18] and patients with disease duration of less than 15 months have demonstrated the best response to high-dose steroid treatment.[19] Recent trials with steroids and IVIG, along with plasmapheresis have proved to be beneficial, especially in adult-onset RE.[18]
To summarize, this is the first reported series of biopsy-proven RE from India, which also emphasizes the cardinal role of histopathology in the confirmation of clinically and radiologically suspected cases. This series also highlights the spectrum of neuropathological findings and characterization of the inflammatory changes, where a trend to inverse correlation of disease duration with degree of inflammation has been observed. Although considerable work has been done[9],[20] to provide a clinical staging system of RE, it is recommended that studies with larger number of cases be undertaken to correlate the neuropathology spectrum with MRI changes, in order to develop a non-invasive method of staging of the disease, since tissue diagnosis is not available in all cases.
REFERENCES
| 1. | Rasmussen T, Olszewski J, Lloyd-Smith D. Focal seizures due to chronic localized encephalitis. Neurology 1958;8:435-45. Back to cited text no. 1 |
| 2. | Topcu M, Turanli G, Aynaci FM, Yalnizoglu D, Saatci I, Yigit A, et al . Rasmussen encephalitis in childhood. Childs Nerv Syst 1999;15:395-402. Back to cited text no. 2 |
| 3. | Bien CG, Urbach H, Deckert M, Schramm J, Wiestler OD, Lassmann H, et al . Diagnosis and staging of Rasmussen's encephalitis by serial MRI and histopathology. Neurology 2002;58:250-7. Back to cited text no. 3 |
| 4. | Robitaille Y. Neuropathologic aspects of chronic encephalitis. In: Andermann F (ed). Chronic encephalitis and epilepsy: Rasmussen's syndrome. Boston: Butterworth-Heinemann;1991. pp 79-110. Back to cited text no. 4 |
| 5. | Gupta PC, Roy S, Tandon PN. Progressive epilepsy due to chronic persistent encephalitis. J Neurol Sci 1974; 22: 105-20. Back to cited text no. 5 |
| 6. | Tobias SM, Robitaille Y, Hickey WF, Rhodes CH, Nordgren R, Andermann F. Bilateral Rasmussen Encephalitis: Postmortem Documentation in a Five- year-old. Epilepsia 2003;44:127-30. Back to cited text no. 6 |
| 7. | Nakasu S, Isozumi T, Yamamoto A, Okada K, Takano T, Nakasu Y. Serial magnetic imaging findings of Rasmussen's encephalitis: case report. Neurol Med Chir Tokyo 1997; 37: 924-8. Back to cited text no. 7 |
| 8. | Honavar M, Meldrum BS. Epilepsy. In: Graham D, Lantos PL, editors. Greenfield's neuropathology. 7th ed. London: Arnold, 2002 Greenfield, pp 909-11. Back to cited text no. 8 |
| 9. | Bien CG, Widman G, Urbach H, Sassen R, Kuczaty S, Wiestler OD, et al . The natural history of Rasmussen's encephalitis. Brain 2002; 125:1751-9. Back to cited text no. 9 |
| 10. | Chiapparini L, Granata T, Farina L, Ciceri E, Erbetta A, Ragona F, et al . Diagnostic imaging in 13 cases of Rasmussen's encephalitis: can early MRI suggest the diagnosis? Neuroradiology 2003; 45:171-83. Back to cited text no. 10 |
| 11. | Pardo CA, Vining EP, Guo L, Skolasky RL, Carson BS, Freeman JM. The pathology of Rasmussen syndrome: stages of cortical involvement and Neuropathological studies in 45 hemispherectomies. Epilepsia 2004; 45: 516-26. Back to cited text no. 11 |
| 12. | Jay V, Becker LE, Otsubo H, et al . Chronic encephalitis and epilepsy (Rasmussen's encephalitis). Detection of cytomegalovirus and herpes simplex virus 1 by the polymerase chain reaction and in situ hybridization. Neurology 1995; 45:108-17. Back to cited text no. 12 |
| 13. | Andrews JM, Thompson JA, Pysher TJ, Walker ML, Hammond ME. Chronic encephalitis, epilepsy, and cerebrovascular immune complex deposits. Ann Neurol 1990; 28: 88-90. Back to cited text no. 13 |
| 14. | Hart YM, Andermann F, Robitaille Y, Laxer KD, Rasmussen T, Davis R. Double pathology in Rasmussen's syndrome: a window on the etiology? Neurology 1998; 50:731-5. Back to cited text no. 14 |
| 15. | Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B, et al . Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis. Science 1994 ;265:648-51. Back to cited text no. 15 |
| 16. | Watson R, Jiang Y, Bermudez I, Houlihan L, Clover L, McKnight K et al . Absence of antibody to glutamate receptor type 3 (Glu R3) in Rasmussen encephalitis. Neurology 2004; 63: 43-50. Back to cited text no. 16 |
| 17. | Bien CG, Bauer J, Deckwerth TL, Wiendl H, Deckert M, Wiestler OD, et al . Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen's encephalitis. Ann Neurol 2002;51:311-8. Back to cited text no. 17 |
| 18. | Bien CG, Elger CE, Wiendl H. Advances in pathogenic concepts and therapeutic agents in Rasmussen's encephalitis. Expert Opin Investig Drugs 2002; 11: 981-9. Back to cited text no. 18 |
| 19. | Chinchilla D, Dulac O, Robain O, Plouin P, Ponsot G, Pinel JF, et al . Reappraisal of Rasmussen's syndrome with special emphasis on treatment with high doses of steroids. J Neurol.Neurosurg.Psychiatry 1994;57:1325-33. Back to cited text no. 19 |
| 20. | Oguni H, Andermann F, Rasmussen TB. The natural history of the syndrome of chronic encephalitis and epilepsy: a study of the MNI series of forty-eight cases. In: Anderman F (ed). Chronic encephalitis and epilepsy: Rasmussen's syndrome. Boston: Butterworth-Heinemann; 1991. p. 7-35. Back to cited text no. 20 |
Copyright 2005 - Neurology India
The following images related to this document are available:
Photo images
[ni05045f2.jpg]
[ni05045t1.jpg]
[ni05045f3.jpg]
[ni05045f5.jpg]
[ni05045f4.jpg]
[ni05045f1.jpg]
|

{kind=link}
![[Figure - 1]](/showimage?ni/photo/ni05045f1.jpg){kind=link}
![[Figure - 2]](/showimage?ni/photo/ni05045f2.jpg){kind=link}
![[Figure - 3]](/showimage?ni/photo/ni05045f3.jpg){kind=link}
![[Figure - 4]](/showimage?ni/photo/ni05045f4.jpg){kind=link}
![[Figure - 5]](/showimage?ni/photo/ni05045f5.jpg){kind=link}