 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
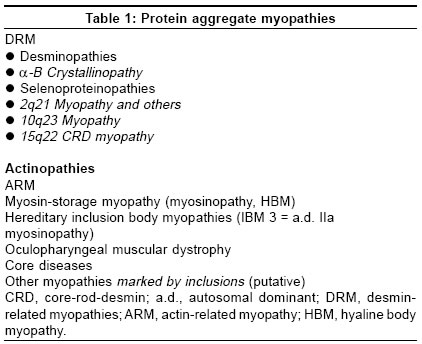
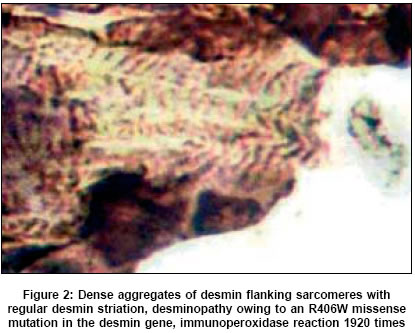
Neurology India, Vol. 53, No. 3, July-September, 2005, pp. 273-279 Review Articles Protein aggregate myopathies Sharma MC, Goebel HH Department of Neuropathology, Johannes Gutenberg University Medical Center, Mainz Date of Acceptance: 08-Oct-2004 Code Number: ni05094 Abstract Protein aggregate myopathies (PAM) are an emerging group of muscle diseases characterized by structural abnormalities. Protein aggregate myopathies are marked by the aggregation of intrinsic proteins within muscle fibers and fall into four major groups or conditions: (1) desmin-related myopathies (DRM) that include desminopathies, a-B crystallinopathies, selenoproteinopathies caused by mutations in the, a-B crystallin and selenoprotein N1 genes, (2) hereditary inclusion body myopathies, several of which have been linked to different chromosomal gene loci, but with as yet unidentified protein product, (3) actinopathies marked by mutations in the sarcomeric ACTA1 gene, and (4) myosinopathy marked by a mutation in the MYH-7 gene. While PAM forms 1 and 2 are probably based on impaired extralysosomal protein degradation, resulting in the accumulation of numerous and diverse proteins (in familial types in addition to respective mutant proteins), PAM forms 3 and 4 may represent anabolic or developmental defects because of preservation of sarcomeres outside of the actin and myosin aggregates and dearth or absence of other proteins in these actin or myosin aggregates, respectively. The pathogenetic principles governing protein aggregation within muscle fibers and subsequent structural sarcomeres are still largely unknown in both the putative catabolic and anabolic forms of PAM. Presence of inclusions and their protein composition in other congenital myopathies such as reducing bodies, cylindrical spirals, tubular aggregates and others await clarification. The hitherto described PAMs were first identified by immunohistochemistry of proteins and subsequently by molecular analysis of their genes. Keywords: actin; aggregation of proteins; congenital myopathies; desmin; inclusion body myopathies; myosin Aggegation of proteins within muscle cells may be of lysosomal or nonlysosomal nature. The former occurs, for instance, in the neuronal ceroid-lipofuscinoses (NCL) as components of lipopigments, their accretion being a morphological hallmark in the NCL.[1] This lysosomal accumulation of lipopigments renders the NCL a member of the lysosomal disorders among which type-II glycogenosis, Niemann-Pick disease, mucolipidosis IV and others may affect the skeletal muscle. Nonlysosomal accumulation of proteins within muscle fibers is a hallmark of a recently emerging subgroup of myopathies, called protein aggregate myopathies (PAM). The cause of protein accretion within muscle fibers - and other cell types of other organs in the protein aggregate disorders - may be (a) impaired extralysosomal degradation of proteins, or (b) abnormal synthesis, (c) integration of proteins in the intracellular structured constituents, e.g. sarcomeres. Proteins may aggregate in soluble, granular and filamentous fashions. At light microscopic level such protein aggregates can hardly be distinguished among the three different states of protein aggregation only the granular and filamentous aggregates may be recognized by electron microscopy while aggregation of soluble proteins may remain invisible and electron microscopic immunohistochemical labeling may not necessarily distinguish between a labeled soluble protein and an artificially misplaced label, (largely gold grains with or without silver enhancement) The mutated proteins probably undergo faulty folding or misfolding[2],[3] and inappropriately expose their hydrophobic surfaces for protein-protein interactions. In contrast to correctly folded proteins, which are either soluble or associated with cell membrane, these misfolded proteins are more insoluble and result in aggregation. These aggregated proteins are less amenable to removal by molecular chaperones and the ubiquitin proteasome degradation pathway, which are the two main protective scavenging pathways of eurkaryotic cells against misfolded proteins. Protein aggregate myopathies may, therefore, belong to the emerging group of protein conformational disorders. Protein aggregation in neuroectodermal cells has been known for a long time and is associated with certain diseases, largely of a neurodegenerative nature Like Alzheimer and prion diseases, Parkinson disease, multiple system atrophy, motor neuron and Pick diseases, Alexander disease and a particular form of myoclonus epilepsy, called neuroserpinosis. Recent biochemical and molecular studies have not only further illustrated protein aggregation, the biochemical components of the protein aggregates and the molecular background of many though not all of them, but also have placed ′protein aggregation′as a central pathogenetic issue in the forefront of research in neurodegenerative disorders. Intracellular protein aggregation occurs in Alexander disease as Rosenthal fibers, secondary due to mutations in the glial fibrillary acidic protein gene, in neuroserpinosis as granular neuroserpin following mutations in the neuroserpin gene, and as Lewy bodies containing a-synuclein and neurofilaments, based on mutant a-synuclein in one of the familial types of Parkinson disease. Alzheimer disease, however, shows intracellular accumulation of proteins as paired helical filaments rich in tau and extracellular accumulation of aggregates of fibrils called amyloid. Extracellular fibrillar protein aggregation of amyloid also occurs in prion diseases owing to the extracellular deposition of abnormal prion proteins. Whether intracellular aggregation of proteins, which are often hyperphosphorylated, follow pathogenetic principles similar or identical to those of extracellular protein aggregation has not completely been clarified. Extracellular protein aggregation, apart from formation of AL-amyloid as a feature of generalized amyloidosis owing to formation of abnormal immunoglobulins in plasma cell dyscrasias and plasmacytoma, has not been seen in any of the myopathies marked by intracellular protein aggregation. Pathogenesis and morphogenesis in protein aggregation within muscle fibers are still incompletely understood. The frequent occurrence of protein aggregation in genetic myopathies, which is responsible for the formation of mutant poteins, suggests that respective mutant proteins play a crucial role in protein aggregation, perhaps acting as seeds for the assembly of other nonmutant proteins as well. Among sarcomeric and sarcomere-related proteins, numerous proteins are associated with certain myopathies, which largely belong to the group of congenital myopathies (CM). Nemaline myopathies are found genetically heterogeneous, involving mutations in genes for nebulin, actin, troponin-T and tropomyosins 2 and 3, of which, however, only actin may accumulate in respective patients with the mutational ACTA 1 type of nemaline myopathy. Although a-actinin is a major protein of the Z-band and present in aggregates of Z-disk material or nemaline bodies/rods, mutations in the respective actinin gene have not been identified as a cause of nemaline myopathy. Titin, telethonin, myotilin, and the enzyme protein calpain-3 give rise to certain muscular dystrophies of the limb-girdle muscular dystrophy series. Myosin and desmin are additional proteins found aggregated in muscle fibers in other PAM. One of the trinucleotide repeat disorders, oculopharyngeal muscular dystrophy, is also marked by intranuclear accumulation of filaments representing the poly (A)-binding protein 2. Thus, the current group of PAM encompasses DRM, actin-related myopathies, myosin-related myopathies and a few others [Table - 1]. PAM suggesting faulty protein degradation The largest subgroup of myopathies falling into this category are those marked by the accumulation of desmin and, as a heterogeneous subgroup by itself, collectively also termed ′myofibrillar myopathy′.[4] This latter term refers to the myofibrillar lesions occurring when desmin accumulates within muscle fibers and, thus, emphasizes the sequelae to protein aggregation and disruption of sarcomeres. Clinically, DRM are often of adult onset and, thereby, deviating from the experience in other CM, which usually commence in childhood. Further distinctive features are the distribution of muscle weakness, with generalized or proximal weakness prevailing in childhood CM but often distal in DRM, and cardiac symptoms, occasionally even resulting in sudden death in DRM. This latter feature of cardiac involvement is not surprising as abnormalities of desmin, the muscle cell-specific intermediate filament protein affects skeletal muscle fibers, cardiac myocytes and smooth muscle cells, i.e. in man by aggregation of desmin, in the desmin knock-out mouse by severe necrotizing lesions in the cells and respective organs such as skeletal muscle, heart and major arteries. Desmin forms an intricate intracellular network or cytoskeleton, linking nuclei via sarcomeric Z-bands to the plasma-sarcolemma. In PAM not only protein aggegation occurs as a conspicuous morphological hallmark but, most likely, also causes disruption of this intermediate filament network and, thereby, its function. Morphologically, DRM may be grouped into two types, those marked by inclusion bodies and those marked by accumulation of granulofilamentous material, although both features have simultaneously been observed. Respective terms such as cytoplasmic bodies, Mallory body-like inclusions or granulofilamentous material, earlier marking certain CM, only entered the group of PAM when immunohistochemistry had been applied to these lesions. [5],[6],[7] The identification of desmin and a multitude of other diverse proteins in these inclusions[8],[9] has brought these once considered separate CM into PAM. Recent molecular studies have further corroborated histological diversity of the DRM, with mutations in the desmin gene being largely though not exclusively of a missense type.[10] Following the principle of re-naming Duchenne-Becker muscular dystrophies as dystrophinopathies and certain autosomal-recessive limb-girdle muscular dystrophies as sarcoglycanopathies, based on mutations in the respective genes encoding dystrophin and sarcoglycans, the term desminopathy appears appropriate for myopathies marked by mutations in the desmin gene. Another group of DRM are actually a-b crystallinopathies, clinically and morphologically identical to desminopathies with cardiomyopathy and granulofilamentous material accruing within muscle fibers, owing to mutations in the small heat-shock or chaperone protein α-β crystallin.[11],[12] Another DRM, recently further elucidated at the molecular level is Mallory body-like myopathy,[13],[14] an autosomal-recessive childhood myopathy marked by scoliosis, muscle weakness and severe respiratory insufficiency which has now been found related to a homozygous mutation in the selenoprotein N1 (SEPN1) genes.[15] Whether, SEPN1 actually accumulates in these lesions is currently unknown because a respective antibody has not yet successfully been applied to these muscle specimens. Selenoproteinopathies not only encompass Mallory body-like myopathy, but also the rigid spine form among the congenital muscular dystrophies[16] and one type of multi-minicore disease.[17]
Further DRM link to other chromosomes, 10q,[18] 2q21,[7],[19] and 15q22 core-rod desmin myopathy.[20] Hereditary inclusion body myopathies This group of disorders may be classified among the PAM because different proteins aggregate within muscle fibers, which contain autophagic vacuoles, and within tubulofilamentous aggregates which are the hallmark of inclusion body myopathies (IBM).[23] Similarities between IBM and DRM are distribution of muscle weakness, occurring later than childhood-onset of clinical symptoms. Autophagic vacuoles, tubulofilamentous aggregates and paired helical filaments have occasionally been seen in DRM.[14],[24],[25] Similar morphological features and a variety of proteins have been identified in IBM also though not disease-specific mutant proteins. A further parallel may exist between genetic and nongenetic DRM as well as hereditary and sporadic, inflammatory IBM. A coμn denominator of the DRM and IBM is probably an impaired, misguided, nonlysosomal protein degradation, the reason of which may easier be explained when dealing with mutant proteins, but still is hardly understood in sporadic acquired DRM and IBM. Protein aggregate myopathies due to possible developmental defects Desmin-related myopathies and IBM, both of hereditary and acquired types, especially when involving mutant proteins may be caused by impaired extralysosomal protein degradation and aggregation while other types of PAM suggest a developmental defect in synthesis, integration and maturation of proteins into fully functional constituents of the muscle fiber. The former group of PAM concerns proteins, which not necessarily are components of sarcomeres while the latter group entails sarcomeric proteins, e.g. actin and myosin. Further evidence to suggest that anabolic or synthetic defects rather than catabolic events in mutational defects of the sarcomeric proteins, actin and myosin are responsible for the frequent occurrence of clinical symptoms in early childhood and often with rapid fatal outcome. In ′catabolic′ PAM, DRM, and IBM many proteins co-aggregate instead of other proteins accumulating inside the muscle. Another feature in the two types of myopathies, actinopathy and myosinopathy is the preservation of sarcomeres close to the accumulating actin filaments and hyaline bodies while in DRM the sarcomeric pattern is severely disturbed, giving rise to the term ′myofibrillar myopathy′.[4],[26]Actinopathies Actin, the major component of thin myofilaments, appears to accumulate within muscle fibers in a two pathomorphological forms: first, as inclusion bodies, (nonspecific filamentous bodies) frequently seen in the subsarcolemmal region by electron microscopy and Hirano bodies, once described in muscle fibers in a nonspecific myopathy[27] similar to Hirano bodies seen in aged neurons of the hippocampus, as another nonspecific morphological phenomenon. The second morphological feature of filamentous actin aggregation appears as plaques, patches, or areas devoid of sarcomeres, particularly but not exclusively situated underneath the sarcolemma, in actin myopathies. Morphologically, actin aggregates are marked by the absence of both oxidative enzyme and adenosine triphosphatase activities in histochemical preparations. The term actinopathy indicates mutations in the sarcomeric ACTA1 gene - similar to the terms dystrophinopathy and sarcoglycanopathies which indicate mutations in the respective genes encoding dystrophins and the sarcoglycans, although evidence for the aggregation of mutant actin has not yet unequivocally been demonstrated. Over 90 mutations in the ACTA1 gene have now been identified in a large number of patients,[28],[29] often being de novo mutations leading to an autosomal-dominant mode of inheritance. Most of them have been missense mutations,[30] while one each of duplication, nonsense mutation and splice site mutation being additionally described.[29] However, patients affected by mutations in the ACTA1 gene by far outnumber of those patients whose muscle biopsy specimens reveal aggregates of actin filaments. On the other hand, nearly all patients with ACTA1 gene mutations also had nemaline bodies in their muscle fibers, thus, qualifying for the label of nemaline myopathy. Therefore, mutations in the ACTA1 gene now belong to the genetic spectrum of nemaline myopathies though not necessarily in all patients with aggregation of actin filaments and ACTA1 gene mutations because some patients did not have any rods at all while others had only intranuclear rods.[28],[31] The precise inter relationship between the accumulation of actin filaments, the sarcoplasmic rods, and intranuclear rods, is still not understood completely. Within the nosological spectrum of nemaline myopathies, mutations in other genes responsible for nemaline myopathies are those encoding other sarcomeric proteins such as tropomyosins 2[32] and 3,[33],[34] troponin[35] and nebulin,[36],[37] suggesting the formation of rods may be a response to miscoded proteins. The clinical spectrum of ACTA1 mutations is as large as that of nemaline myopathies in general, although, patients with ACTA1 mutations in particular seem to suffer more often from a severe early childhood form compared to patients with mutations in the nebulin gene. Similarly a broad clinical spectrum can be seen in the subgroup of these ACTA1 mutations where several children suffering from actinopathies marked by actin aggregates showed a severe and rapidly fatal course while others had a much milder condition.[28],[29],[31] The patient reported by Bornemann[38] who succumbed at 5 months of age, had a duplication.[29] The type of ACTA1 mutation does not seem to have any effect on the outcome of the disease as the same Val163Leu miscoded actin protein was found in a rapidly deceased child as well as in a mildly affected patient, allowing a near-normal adolescence with little muscle weakness.[28],[29],[31] Cardiac symptoms, cardiomyopathy, and untimely cardiac death are not features of actinopathy because cardiac actin differs genetically from sarcomeric actin, the mutations of the former at locus 15q14 giving rise to separate cardiomyopathies. The most recent observation in a floppy infant with severe respiratory distress includes large aggregates of actin within muscle fibers but no mutations in the ACTA1 gene, justify the term actin-related myopathy (ARM), similar to the term DRM when mutations in the desmin gene were not present or were excluded. However, mutations in other genes have not yet been identified in this patient.[39] Myosinopathy Myosinopathy is the most recent myopathy from the premolecular era added to this group of PAM. Myosinopathy, so far, marked by an Arg1845Trp mutation at locus 14q11.2 in the slow/β -cardiac myosin heavy chain gene,[40] had earlier been described first as myofibrillar lysis myopathy[41] and later as hyaline body myopathy (HBM).[42],[43] Clinically, HBM has been recorded both in children, their siblings,[41] and in adults.[40],[42],[44],[45] The appearance in siblings[41],[46] suggested an autosomal-recessive mode of inheritance while an autosomal-dominant form has also been described.[44] The autosomal-recessive form seems to run a more rapid and even fatal course as one of the two original siblings reported by Cancilla et al.[41] died in her 30 s (B. Azzarelli, Indianapolis: personal communication). Morphologically, hyaline bodies are actually not bodies but inclusions of the plaque or patch type, devoid of oxidative enzyme activities though not of ATPase activities. Structurally, these plaques consist of finely granular material without any further structures affected. The plaques of granular material in hyaline bodies appear sharply demarcated from surrounding intact sarcomeres. Immunohistochemically, these plaques are not only rich in myosin[42],[44] but are devoid of any other proteins tested,[42],[44] suggesting that the accumulating granular material is actually an ATPase-positive myosin protein. Thus, it did not come as a surprise that mutational analysis of the sarcomeric myosin protein detected an Arg1845Trp mutation in the myosin heavy chain gene MYH-7of respective patients.[40] Although mutations in the MYH-7 gene, responsible for myosinopathy, have also been seen in certain cardiomyopathies, cardiac symptoms, cardiomyopathy or immature cardiac death do not seem to be nosological features of myosinopathy. Recently, another gene locus 3p22.2-21.32 has also been linked to autosomal-recessive HBM.[47] Other possible PAM - core diseases Core diseases, central core disease (CCD) and multiminicore disease (MmD), are marked by focal destruction of sarcomeres, located centrally within muscle fibers in CCD and multifocally across the muscle fiber in MmD. These areas are also found to be devoid of mitochondria, allowing recognition of both central and multiminicores in oxidative enzyme preparations, a morphological hallmark of core diseases. It has been known for some time that different proteins aggregate in nonstructured cores[48] and in multiminicores.[49] As regards central cores, this aggregation of proteins has been seen in nonstructured cores and in targets and targetoid lesion following denervation[48] which suggests accumulation of proteins as a secondary event, either because of the core and targetoid lesions or because of the absence of mitochondria, or both. As CCD is often based on mutations in the Ryanodine receptor 1 gene (RYR1), accumulation of the RYR1 protein in such cores in CCD[50] suggests, deposition of a mutant RYR1 protein in central cores as a possible seed for the subsequent accumulation of other proteins. This, then, may not necessarily be a secondary event but a primary lesion. The structured central cores with preservation of sarcomeres, though different extracore regions in the same muscle fiber, by the absence of mitochondria resulting in absence of reaction substrate in oxidative enzyme preparations, whether aggregation of proteins included a putative mutant RYR1 protein in CCD with structured central cores or CCD with both structured and unstructured cores, remains to be clarified. Multiminicore disease has now genetically been found heterogeneous, one form representing an early stage of CCD[51] and another is marked by mutations in the SEPN1 gene.[17] Whether mutant RYR1 protein aggregating in the CCD form of MmD or mutant SEPN1 in the selenoproteinopathy type of MmD, has not yet been elucidated. Unequivocal ′structured′multiminicores have not convincingly been demonstrated. Future perspectives Evolution of the PAM concept originated from morphological observations on accumulation and aggregation of proteins in the muscle fiber like desmin, actin, myosin, though later often thought to be mutants ′from protein to mutation′. This has prompted molecular studies on the respective genes, which have now provided a wealth of information about these genes and considerably advanced our knowledge on this group of CM marked by structural abnormalities, the ′protein aggregating myopathies′.Future investigations will have to address
Acknowledgments We are grateful to skilled assistance by Astrid W φber (editing) and Walther Wagner (illustrations).References
Copyright 2005 - Neurology India The following images related to this document are available:Photo images[ni05094t1.jpg] [ni05094f4.jpg] [ni05094f3.jpg] [ni05094f1.jpg] [ni05094f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}