 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
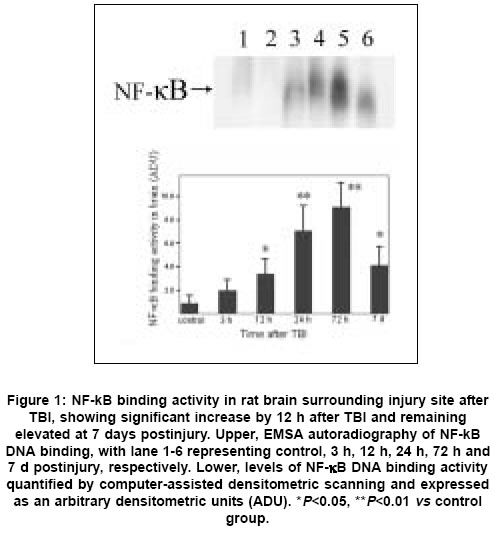
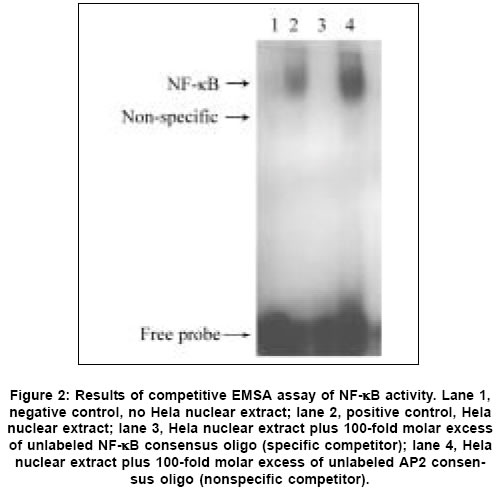
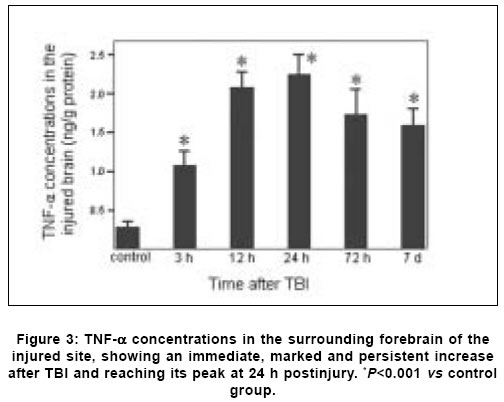
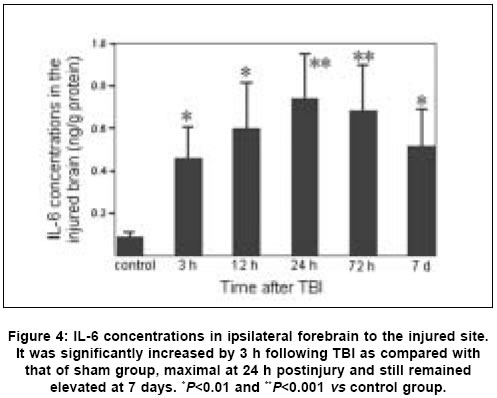
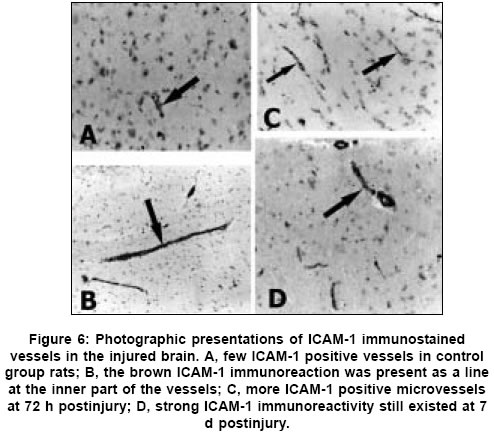
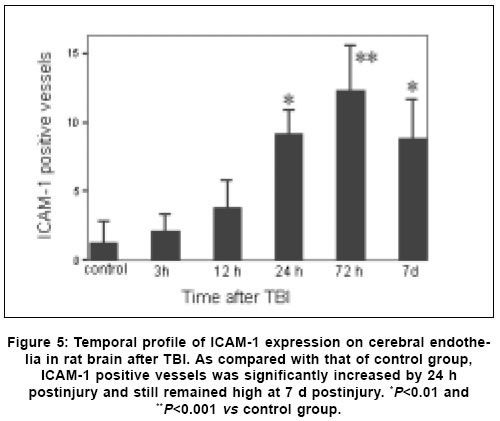
Neurology India, Vol. 53, No. 3, July-September, 2005, pp. 312-317 Original Article Concomitant upregulation of nuclear factor-kB activity, proinflammatory cytokines and ICAM-1 in the injured brain after cortical contusion trauma in a rat model Hang ChunHua, Shi Ji-Xin, Li Jie-Shou, Wu Wei, Yin HongXia Department of Neurosurgery, Jinling Hospital, Clinical School of Medicine, Nanjing University, Nanjing 210002 Date of Acceptance: 03-May-2005 Code Number: ni05110 Abstract Background: Nuclear factor kappa B (NF-κB), proinflammatory cytokines and intercellular adhesion molecule 1 (ICAM-1) are frequently upregulated in the injured brain after traumatic brain injury (TBI). However, the temporal pattern of upregulation is not well defined.Aims: The current study was undertaken to investigate the temporal profile of the expression of NF-κB, proinflammatory cytokines and ICAM-1 in the injured brain after cortical contusion trauma of the rat brain. Settings and Design: A rat model of cortical contusion was produced by a free-falling weight on the exposed dura of right parietal lobe. The rats were randomly divided into control group and TBI groups at hours 3, 12, 24 and 72, and on day 7. Material and Methods: NF-κB binding activity in the surrounding brain of injured area was studied by electrophoretic mobility shift assay (EMSA). The levels of TNF-α and IL-6 were detected using ELISA and ICAM-1 expression studied by immunohistochemistry. Statistical analysis: The data were analyzed by one-way ANOVA followed by Student-Newman-Keuls post hoc test. Relation between variables was analyzed using bivariate correlation with two-tailed test. Results: Compared with that of control group, NF-κB binding activity in the injured brain was significantly increased through 12 h and 7 days postinjury, with the maximum at 72 h. The concentrations of TNF-α and IL-6 in the injured brain were significantly increased from 3 h to 7 days and maximal at 24 h postinjury. The number of ICAM-1 immunostained microvessels was significantly increased in the injured brain from 24 h to 7 days postinjury, with its peak at 72 h. Concomitant upregulation of TNF-α, IL-6, ICAM-1 and the cytokine mediators NF-κB in the injured brain was observed in the injured brain after cortical contusion, and there was a highly positive relation among these variables. Conclusions: Cortical contusion trauma could induce a concomitant and persistent upregulation of NF-κB binding activity, TNF-α, IL-6 and ICAM-1 in the injured rat brain which might play a central role in the injury-induced immune response of brain. Keywords: Traumatic brain injury; Nuclear factor-kB; Proinflammatory cytokine; Intercellular adhesion molecule-1; inflammation. Introduction Several reports from clinical and experimental studies have demonstrated that secondary brain injury can be magnified by numerous immune mediators which are frequently upregulated in response to traumatic brain injury (TBI). [1],[2],[3],[4],[5] Increased levels of these molecules within the injured brain, including TNF-α, IL-6 and intercellular adhesion molecule 1 (ICAM-1), are believed to contribute to the cerebral damage, cell death and blood-brain-barrier (BBB) dysfunction.[6] A pivotal player in the regulation of these molecules is the nuclear factor kappa B (NF-κB) family (cRel, RelA/p65, Rel B, p50 and p52) of transcription factors.[7]-[8] NF-κB can be activated by lesion-induced oxidative stress, bacterial endotoxin or cytokines, and subsequently activate transcriptionally the genes encoding cytokines, prostaglandin synthase-2, ICAM-1 and inducible nitric oxide synthase (iNOS). Although it was not present in nondegenerating neurons,[10] upregulation of NF-κB binding activity was detected in the cerebral cortex ipsilateral to the injury site following TBI. [11],[12],[13] However, discrepancy existed among those studies on the temporal pattern of upregulation of NF-κB binding activity, proinflammatory cytokines and ICAM-1, i.e., early versus delayed. Till now, no study was found in the literature to simultaneously investigate the NF-κB binding activity, proinflammatory cytokine and ICAM-1 expression in a same TBI rat model. It is not well known about the difference in temporal profile of upregulation of NF-κB and these immune molecules within the injured brain following TBI. We, therefore, used a weight-drop model of rats to mimic a cortical contusion trauma and to study the temporal profile of upregulation of the NF-κB binding activity, proinflammatory cytokine and ICAM-1 in the injured brain. Material and methods Male Wistar rats (220g to 250g) were purchased from Animal Center of Chinese Academy of Sciences, Shanghai, China. The rats were housed in temperature- and humidity- controlled animal quarters with a 12 hour light/dark cycle, room temperature at 23 ± 1°C and free access to food and water ad lib . All procedures were approved by the Institutional Animal Care Committee, and were in accordance with the guidelines of the National Institutes of Health on the care and use of animals. Rat model of cortical contusion trauma: The rats were randomly divided into six groups (6 rats/group) including control group with right parietal bone window alone and no brain injury, and TBI groups at hours 3, 12, 24, 72 and days 7, respectively. Following intraperitoneal anesthesia with urethane (1000 mg/kg), animal head was fixed in the stereotactic frame. A right parietal craniotomy (diameter 5mm) was drilled under aseptic conditions 1 mm posterior and 2 mm lateral to the bregma. We used a modification of the Feeney′s weight-drop model[14] in which a freefalling weight onto the exposed intact cranial dura produced a standardized parietal contusion by letting a steel rod weighing 40 g with a flat end diameter of 4 mm fall onto a piston resting on the dura from a height of 25 cm. The piston was allowed to compress the tissue a maximum of 5 mm. Animals of control group were killed for sample collection at 72 h after sham injury. TBI group rats were decapitated at corresponding time point postinjury. The ipsilateral forebrain tissue adjacent to the injured cortex was dissected on ice, some of which was put into 10% buffered formalin; the other was stored at liquid nitrogen immediately until use. Nuclear protein extracts and EMSA: NF-κB was examined using electrophoretic mobility shift assay (EMSA). Nuclear extracts were prepared as previously described in detail.[15] The injured or sham injured cerebral cortex ipsilateral to injury site was homogenized with 1 ml of a buffer composed of 10 mM HEPES (pH 7.9), 2 mM MgCl2, 10 mM KCl2, 0.1 mMEDTA, 1 mM dithiothreitol (DTT) and 0.5 mM phenylmethylsulfonyl fluoride (PMSF) (all from Sigma Chemical Co.). The homogenates were centrifuged for 30 s at 1000g at 4°C to eliminate any unbroken tissue. The supernatants were incubated on ice for 20 min and centrifuged at 5000g at 4°C for 10 min. The crude nuclear pellets were suspended in 200 µl ice-cold buffer containing 20 mM HEPES (pH 7.9), 25% (v/v) glycerol, 1.5 mM MgCl2, 20 mM KCl, 0.1 mM EDTA, 0.5 mM PMSF, and 1 mMDTT and incubated on ice for 30 min, mixed frequently, and centrifuged at 12,000g at 4°C for 15 min. The supernatants were collected as nuclear extracts and stored at -80°C for further analysis. Protein concentration was determined using a bicinchoninic acid assay kit with bovine serum albumin as the standard (Pierce Biochemicals, Rockford, IL). EMSA was performed using a commercial kit (Gel Shift Assay System, Promega, Madison, WI). The NF-κB consensus oligonucleotide probe (5′-AGTTGAGGGGAC-TTTCCCAGGC-3′) was end-labeled with [g-32subP] ATP (Free Biotech, Beijing, China) with T4-polynucleotide kinase. Nuclear protein (40 µg) was preincubated in a total volume of 9 µl in a binding buffer, consisting of 10 mM Tris-HCl (pH 7.5), 4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, and 0.05 mg/ml poly (di-dc) for 10 min at room temperature. After addition of the32sub P-labeled oligonucleotide probe, the incubation was continued for 20 min at room temperature. The specificity of the DNA/protein was determined by competition reactions in which a 100-fold molar excess of unlabeled NF-κB oligonucleotide (specific competitor) or unlabeled AP2 oligonucleotide (nonspecific competitor) was added to the binding reaction 10 min before the addition of radiolabeled probe using Hela nuclear extract. Reaction was stopped by adding 1 µl of gel loading buffer and the mixture was subjected to nondenaturing 4% polyacrylamide gel electrophoresis in a TBE buffer (Tris-borate-EDTA). The gel was vacuum-dried and exposed to X-ray (Fuji Hyperfilm) at -70°C with an intensifying screen. Detection of TNF-α and IL-6 in the injured brain: Detection of TNF-α and IL-6 in the injured brain was made using enzyme linked immunoabsorbant assay (ELISA). The frozen brain tissue was homogenized with a glass homogenizer in 1 ml buffer containing 1μl/l phenylmethylsulfon glfluoride (PMSF), 1mg/l pepstatin A, 1 mg/l aprotinin, and 1mg/l leupeptin in phosphate-buffered saline solution (pH 7.2) and centrifuged at 12000 g for 20 min at 4 °C. The supernatant was then collected and total protein was determined by Bradford method. The levels of TNF-α in the tissue supernatants were measured using an ELISA kit (Diaclone, France) specific for rat TNF-α. IL-6 levels in the tissue supernatants were determined with a rat ELISA kit (Biosource, USA). The measurement of both TNF-α and IL-6 was performed step by step based on the protocol booklet of ELISA kit. According to the specifications given by manufacturer, the inter-assay coefficient and intra-assay coefficient of variation for TNF-α were 7.9%-8.2% and 6.1%-6.5%, respectively. The minimum limit of TNF-α detection for this assay was 20pg/ml. Intra- and inter-assay coefficients of variation for IL-6 were 3.7%-4.9% and 5.9%-9.9%, respectively, with the detection limit for this assay less than 8pg/ml. The cytokine contents in the injured brain were expressed as ng cytokines/g protein. Detection of ICAM-1 expression in the injured brain: Detection of ICAM-1 expression in the injured brain was made by immunohistochemistry. The 10% buffered formalin-fixed brain tissue was embedded in paraffin, sectioned at 4 m thickness with a microtome and stained with hematoxylin and eosin (H-E). The H-E stained sections were examined under microscope for any alteration in histopathology. Rabbit Anti CD54 (ICAM-1) primary antibody was purchased from Boster Biotechnology Co.Ltd, China and the working dilution for these antibodies was 1:100. For immunohistochemistry, sections were incubated in phosphate-buffered saline (PBS) with 5% normal horse serum and 0.3% Triton X-100 for 1 h at room temperature (RT). sections were washed three times with PBS and incubated with primary antibody to ICAM-1 for 2 h at RT. After being washed with PBS, sections were incubated with biotinylated second antibodies for 1 h at RT. sections incubated in the absence of primary antibody were used as negative controls. Microscopy of the immunohistochemically stained tissue sections was performed by a pathologist blinded to the experimental condition. The number of ICAM-1 immunoreactive microvessels in each section was counted in 10 microscopic fields (at 100 magnifications) throughout identical regions of the injured and the sham-injured brain, and averaged for the number of per visual field. The increase in the number of immunostained microvessels was assessed by comparing the number of labeled vascular profiles per visual field. Statistical analysis: SPSS 10.0 was used for statistical analysis of the data. All data of NF-κB binding activity, cytokine concentrations and ICAM-1 expression were presented as the mean ± SD. The data were analyzed by one-way anova followed by Student-Newman-Keuls post hoc test. Relation between variables was analyzed using bivariate correlation with two-tailed test. The significance level was set at P <0.05. Results NF-κB binding activity in the injured brain: There was a weak NF-κB activation in the brain of rats with sham injury and cortical contusion trauma for 3 h. As compared with that of control group, NF-κB binding activity in the brain adjacent to the injured site was significantly increased by 12 h after cortical contusion trauma ( P <0.05), was highest at 72 h postinjury, remained elevated at 7 days postinjury [Figure - 1]. The specificity of the shifted bands in EMSA was verified by a competition assay. All the shifted bands were suppressed by incubation with 100-fold excess of unlabeled NF-κB probe and were unchanged by competition with a similar amount of another irrelevant AP2 oligonucleotide [Figure - 2]. The result showed that cortical contusion trauma could induce significant activation of NF-κB within the injured brain as early as 12 h postinjury, which might remain elevated for 7 days. Concentrations of TNF-α and IL-6 in the injured brain: The concentrations of TNF-α and IL-6 were low in the sham-operated rat brain (0.288 ± 0.064 and 0.0895 ± 0.0238 ng/g protein, respectively). As shown in [Figure - 3] and [Figure - 4], concentrations of TNF-α and IL-6 in the injured brain were significantly increased by 3 h after TBI (1.088 ± 0.179 and 0.459 ± 0.147 ng/g protein, respectively) as compared with that of control group, with a maximum at 24 h postinjury (2.254 ± 0.251 and 0.742 ± 0.207 ng/g protein, respectively), and remained significantly elevated by 7 d postinjury (1.591 ± 0.213 and 0.517 ± 0.172 ng/g protein, respectively). Early and persistent upregulation of proinflammatory cytokines, i.e., TNF-α and IL-6, was found in the injured brain, which highly coincided in temporal profile with that of NF-κB activation. There was a positive relationship between NF-κB binding activity and TNF-α level ( r =0.6, P <0.001), between NF-κB binding activity and IL-6 ( r =0.62, P <0.001). ICAM-1 expression in cerebral microvessels: Immunohistochemical study showed that epithelial cells of cerebral vessels in the injured brain displayed increased ICAM-1 immunoreactivity after cortical contusion. The brown immunoreaction was present as a line at the inner part of the vessels, i.e., at the position of the endothelial cell layer [Figure - 6]B. The vast majority of these vessels were of capillary size [Figure - 6]C. Few ICAM-1 immunostained cerebral microvessels were observed in rat brains of control group and TBI 3 h group [Figure - 6]A, showing no any significant difference between these two groups. Compared with that of control group, all the rats after cortical contusion from 24 h to 7 d showed a significant increase in the number of immunostained microvessels in the brain adjacent to the injured site [Figure - 5]. Maximal induction of ICAM-1 immunoreactivity in the cerebrovascular epithelia throughout the lesion was observed at 72 h after cortical contusion trauma [Figure - 6]C and remained elevated at 7 d postinjury [Figure - 6]D. There was a positive relationship between NF-κB binding activity and number of ICAM-1 immunostained microvessels ( r =0.748, P <0.001) in the injured brain after cortical contusion trauma. Discussion In this study, we have reported that concomitant upregulation of the proinflammatory cytokine TNF-α and IL-6, ICAM-1, and their mediators NF-κB occurred in the injured brain after traumatic brain injury, and there was a highly positive relationship among these variables. NF-κB, TNF-α, IL-6 and ICAM-1 were upregulated in the injured brain at early stage of TBI, no less than 24 h postinjury, and remained elevated for several days. It is highly suggested that an interaction exist among NF-κB, proinflammatory cytokines and ICAM-1, mainly through a feedback regulation of complex network.[16] In 1995, Yang et al. first reported the activation of NF-κB in the injured brain following TBI.[11] They demonstrated that NF-κB was activated in the injured cortex on days 3 to 7 after trauma in a cortical impact injury model. Other studies have shown that trauma to the adult brain cortex or spinal cord induces neuronal NF-κB activation between 1 and 7 days postinjury, but not at earlier time point, with a maximum at 3 days. [11],[12],[13] Our results suggested that NF-κB binding activity was significantly increased in the injured brain as early as 12 h, reached its maximum at 72 h and remained elevated at 7 d after TBI. The difference in temporal profile of initially significant upregulation of NF-κB binding activity after TBI may lie in that previous experimental studies had no detecting time point through 12 h and 24 h postinjury. An important mediator in the transcriptional regulation of cytokine gene expression is the NF-κB family of transcription factors. Although typical NF-κB activation is transient, under specific physiological or pathological conditions, inducers such as TNF-α and IL-1 can cause a persistent activation of NF-κB.[17],[18] Activation of NF-κB in the brain is mainly related to the neuronal function[16],[19] and inflammatory response[20] through induction of expression of proinflammatory genes such as TNF-α, IL-1a, IL-6, cyclooxygenase-2 (Cox-2) and ICAM-1.[8] Cerebral upregulation of TNF-α, IL-1 and IL-6 has been evidenced in both clinical and experimental TBI. However, there is some controversy regarding the temporal pattern of upregulation of these molecules. Previous experimental studies after fluid-percussion injury have demonstrated an early increase and maximal levels of the activities of inflammatory cytokines TNF-α and IL-1a within 8 hours after injury.[21] Conversely, Holmin et al.[3] reported the delayed production of cytokines TNF-α and IL-1a (maximally on day 6) in mononuclear cells and reactive astrocytes surrounding the contusion, in contrast to the early expression of these cytokines, which was well correlated with the occurrence of tissue edema. This study showed that both TNF-α and IL-6 level in the injured brain tissue was markedly upregulated from 3 h to 7 d postinjury, with the maximum at 24 h following TBI, which showed a positive relation with the NF-κB activation. The early increase of these cytokines may attribute to the synthesis by nervous cells mainly regulated by activated NF-κB, rather than by infiltrating leukocytes.[1] ICAM-1 is a member of the immunoglobulin supergene family which can be expressed by many cells in the body including endothelial cells. There is a low expression of ICAM-1 in normal brain, which is inducible by tissue injury and inflammatory cytokines such as IL-1 and TNF-α. Up-regulation of ICAM-1 in cerebral microvessels has been described in several pathological conditions of the brain, including brain trauma[22] and cerebral ischemia.[23] In this study, increased ICAM-1 immunoreactivity in the endothelial cells of cerebral microvessels was also observed at the site of lesion after cortical contusion trauma, which showed a positive relation with the NF-κB binding activity. The upregulation of ICAM-1, therefore, coincided in time with the period at which NF-κB binding activity and levels of TNF-α and IL-6 were significantly increased. The functional importance of NF-κB in acute inflammation is based on its ability to regulate the promoters of a variety of genes whose products, such as cytokines, adhesion molecules and acute phase proteins, are critical to inflammatory processes.[7],[24] The inhibition of NF-κB activation by corticosteroid hormones, antioxidants, protease inhibitors and other compounds may provide a pharmacological basis for interference with pathological inflammatory conditions.[25] Sullivan et al.[18] reported that after TBI both the lesion volume and breakdown of the blood brain barrier (BBB) were significantly greater in TNF-receptor knockout (TNFR-KO) mice than in wild-type. Additionally, NF-κB activation was delayed after TBI in TNFR-KO mice, suggesting that TNFR-mediated NF-κB activation may initiate neuroprotective pathways early in the injury process, perhaps via the induction of manganese superoxide dismutase (MnSOD). It is therefore difficult to predict the results of pharmacological intervention with cytokine synthesis after brain injury. The net effects of several attempts, however, are encouraging. A nonspecific inhibition of the inflammatory response can reduce the delayed edema after experimental contusion[26] and the area of tissue damage after blunt trauma.[27] Systemic administration of ICAM-1 antibodies can reduce the number of leukocytes in the parenchyma after reversible cerebral ischemia.[28] In conclusion, we found an early, concomitant and persistent upregulation of NF-κB binding activity, TNF-α, IL-6 and ICAM-1 expression in the injured rat brain after cortical contusion trauma. These results suggest that NF-κB, proinflammatory cytokines and ICAM-1 may play a central role in the injury-induced immune response that leads to secondary insults or reparative processes. A further understanding of the specific role of NF-κB, proinflammatory cytokines and ICAM-1 in brain injury may result in therapy that minimizes secondary brain damage. Acknowledgement We would like to thank Dr. Genbao Feng and Bo Wu for their technical assistance.References
Copyright 2005 - Neurology India The following images related to this document are available:Photo images[ni05110f4.jpg] [ni05110f1.jpg] [ni05110f5.jpg] [ni05110f2.jpg] [ni05110f6.jpg] [ni05110f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}