 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
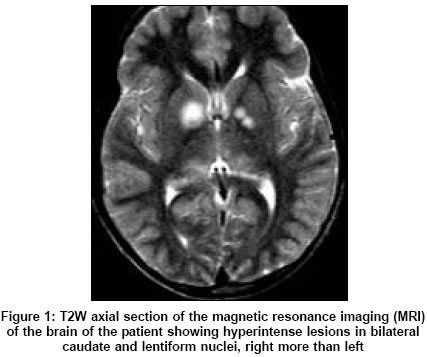
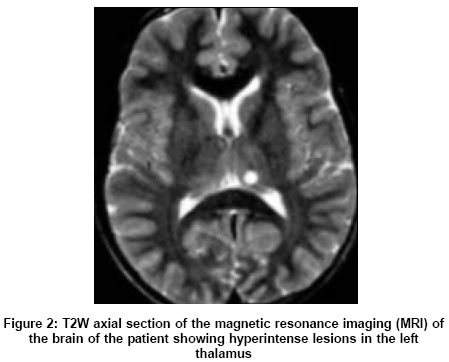
Neurology India, Vol. 53, No. 3, July-September, 2005, pp. 361-362 Letter To Editor Neurofibromatosis type-1 manifesting with Tourette syndrome Kumar Sudhir Department of Neurological Sciences, Christian Medical College, Vellore, Tamilnadu - 632 004 Date of Acceptance: 26-Aug-2004 Code Number: ni05127 Sir, Tourette syndrome (TS) is an inherited neurological disorder with onset below 18 years of age characterized by motor and vocal tics, occurring in 0.6-3% of schoolchildren.[1] The structural basis of TS is incompletely understood. The TS occurring in association with Neurofibromatosis type-1 (NF-1) is a rare feature.[2] However, such a case might provide useful insights into the structural basis of clinical features in TS. A 12-year-old boy presented with multiple motor tics since the age of 5, involving the neck, trunk, upper limbs, and eyelids. The symptoms included eye blinking, arm thrusting, kicking, shoulder shrugging, and jumping. Repeated throat clearing was noted for 18 months. There was no history of seizure, behavioral change, or poor scholastic performance. There were no features to suggest an associated obsessive-compulsive disorder (OCD) or attention deficit hyperactivity disorder (ADHD). There was no similar family history. On examination, multiple cafι-au-lait macules and axillary freckling were present. Neurological examination was unremarkable. Magnetic resonance imaging (MRI) of the brain showed T2W hyperintense lesions in bilateral basal ganglia [Figure - 1] and thalami [Figure - 2]. Electroencephalography was normal. The erythrocyte sedimentation rate was 19 mm/h. Serum ceruloplasmin was 96 mg% (normal > 30 mg%). Antinuclear antibody was negative. Serum complement was 80% and antistreptolysin O titer was normal. The TS was diagnosed on the basis of established clinical criteria. The child showed significant improvement with risperidone 1 mg, taken twice daily. However, MRI abnormalities persisted even after 6 months. On the basis of the etiology, TS is classified into two categories: idiopathic and symptomatic. Symptomatic TS can occur after head injury, encephalitis, stroke, rheumatic fever, and drug intake (methylphenidate, neuroleptics, opioids, antiepileptics). The neuroanatomical localization of tics has remained a challenging feature. Given the wide clinical heterogeneity in TS and frequent association with OCD or ADHD, several neuroanatomical localizations have been evoked, including the cortex (frontal or temporal lobe), limbic system, basal ganglia, and brain stem. The most favored are the striato-thalamic circuits and striatal compartments.[3] The MRI studies in TS have shown significant reduction in the volumes of caudate and lentiform nucleus. Our patient too had involvement of bilateral caudate and lentiform nuclei [Figure - 1]. Therefore, one can speculate that any neurological disease with a significant involvement of caudate or lentiform nuclei or both could present with secondary TS. This argument was strengthened by a recent case of varicella zoster encephalitis with basal ganglia imaging abnormalities, where the patient developed a chronic tic disorder associated with ADHD.[4] In our patient, the basal ganglia abnormalities were related to NF-1, a feature that has been well described.[5] Hyperintense lesions in the basal ganglia in patients with NF-1, may regress over time in 40% of patients, and this might suggest that these lesions are due to demyelination.[5] In conclusion, TS can result from a variety of causes, NF-1 being an uncoμn cause. Our case and data from previous reports point towards the pathological involvement of caudate and lentiform nuclei in the genesis of tics in TS. Tics in secondary TS too respond well to risperidone. References
Copyright 2005 - Neurology India The following images related to this document are available:Photo images[ni05127f2.jpg] [ni05127f1.jpg] |
| |||||||||

{kind=link}
{kind=link}