 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
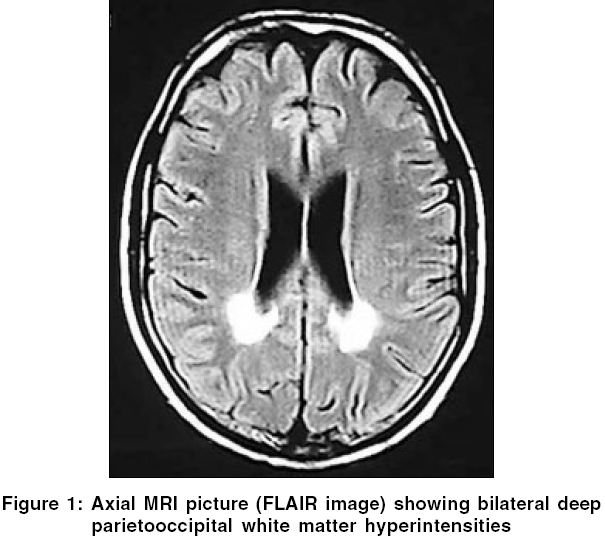
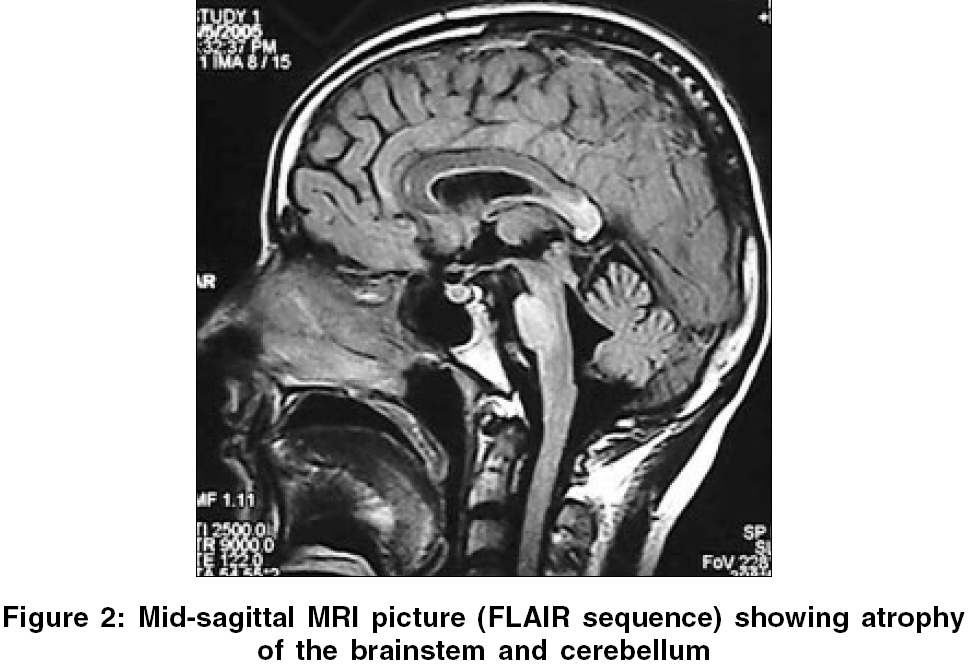
Neurology India, Vol. 54, No. 2, April-June, 2006, pp. 195-196 Case Report Adrenoleukodystrophy manifesting as spinocerebellar degeneration Mishra Sanjay, Modi Manish, Das ChandiP, Prabhakar Sudesh Department of Neurology, PGIMER, Chandigarh - 160 012, Punjab Code Number: ni06057 Abstract X-linked adrenoleukodystrophy (XALD) is an inherited disorder of peroxisomal metabolism. Atypical presentations have been occasionally reported in literature. However, extrapyramidal and cerebellar manifestations are distinctly rare. We report a patient of X-linked adrenoleukodystrophy with cranial and cervical dystonia and neurological presentation resembling spinocerebellar degeneration followed by a brief review of relevant literature.Keywords: Adrenoleukodystrophy, extrapyramidal, spinocerebellar degeneration X-linked adrenoleukodystrophy (XALD) is a rare inherited peroxisomal disorder. Six phenotypes of the disease have been described.[1] The most rapidly progressive childhood cerebral type is the most common followed by gradually evolving adrenomyeloneuropathy.[1] Extrapyramidal manifestation is very rarely reported in patients with XALD. We report a patient of adrenoleukodystrophy having dystonia and clinical presentation resembling spinocerebellar degeneration. Case Report A 29 year old male presented with history of progressive darkening of complexion for 20 years. Four years ago, he was investigated and was found to have low serum cortisol-2.4 μg/dl (normal-5 to 25 μg/dl) and very high ACTH values-6390 pg/ml (normal < 46.00 pg/ml). He was diagnosed to have Addison′s disease and was started on steroid replacement therapy (prednisolone 5.0 mg morning and 2.5 mg evening with fludrocortisone 0.1 mg/day) resulting in dramatic improvement in skin hyperpigmentation. For the last six years he started having hesitancy and later developed increased urinary frequency, urgency and urge incontinence. For the last four years he also had episodes of fecal incontinence and erectile dysfunction. He developed broad based ataxic gait, slurred speech with mild tremulousness while writing for the last two and a half years. Parents also noticed that he became more talkative and argumentative. There was no history of decreased vision/hearing, focal weakness, decreased sensations or seizures. His birth and development was normal. One of his brothers had died at the age of three years with acute gastroenteritis like illness. Examination revealed features of dysautonomia on autonomic testing (orthostatic hypotension, inadequate heart rate variability with deep breathing with inadequate rise in blood pressure on cold pressor and mental arithmetic testing). MMSE score was 30/30. Detailed higher function testing did not reveal any significant abnormality. Speech was ataxic with a spastic component. He had broken pursuits with horizontal gaze evoked nystagmus. Release signs including palmomental reflex, pout and glabellar tap were present. There were bilateral pyramidal (hyperreflexia and extensor plantars) and cerebellar signs. In addition, he had cervical dystonia (antecollis) and cranial dystonia (frontalis/orbicularis dystonia). Investigations revealed low serum cortisol, normal serum luteinizing hormone, follicle-stimulating hormone, testosterone and thyroid hormone levels. Nerve conduction study showed normal latency, amplitude and conduction velocity of motor and sensory nerves. Visual evoked potentials were normal. Brainstem auditory evoked response showed prolonged interpeak latency between I-III and I-V waves on both the sides. Contrast enhanced computed tomography (CT) of the abdomen showed atrophy of both adrenal glands. Magnetic resonance imaging (MRI) of the brain showed hyperintensities on T2 weighted images and FLAIR sequences in bilateral posterior parietooccipital white matter, splenium of corpus callosum, cerebellar white matter, internal capsules, middle cerebellar peduncles, mid brain and pons and marked atrophy of the brainstem and cerebellum [Figure - 1][Figure - 2]. MRI of the cervicodorsal spine showed atrophy of the dorsal cord with hyperintense signals on axial dorsal cuts. Plasma very long chain fatty acids (VLCFAs), C26:0 (0.830 mg/ml) and ratios of C26:0/C22:0 (0.069) and C24:0/C22:0 (1.416) were elevated. (normal values of C26:0 - 0.23±0.09 mg/ml, C26:0/C22:0 - 0.01± 0.004 and C24:0/C22:0 - 0.84±0.10, Sir Ganga Ram Hospital, New Delhi, India). With these clinical and investigation findings diagnosis of adrenoleukodystrophy was made and patient was continued on steroid replacement therapy. Patient could not afford mutational analysis. We wish to report this patient because of several peculiar features like predominant cerebellar and autonomic manifestations, extrapyramidal features and onset with Addison′s disease only phenotype with slow evolution to spinocerebellar degeneration like presentation. Discussion Adrenoleukodystrophy is a disorder of peroxisomal metabolism. The gene (ABCD1) responsible for the disease is located on Xq28. This gene codes for a peroxisomal membrane protein, which facilitates beta-oxidation of VLCFAs. Defect in the gene leads to abnormal membrane protein and thus defective degradation of VLCFAs, which are responsible for the disease.[2] Adrenoleukodystrophy is an important cause of Addison′s disease (4 to 63% in various series), specially in young males.[2] Upto 14% patients of XALD may initially present as Addison′s disease only phenotype and gradually evolve into one or the other phenotype, usually adrenomyeloneuropathy.[3]Besides, six usual phenotypes there are reports of several atypical variants. Rare forms of presentations might mimic spinocerebellar degeneration,[4] olivopontocerebellar atrophy[5] or a brain tumor. Our case presented with cerebellar, autonomic, pyramidal and extrapyramidal features resembling spinocerebellar degeneration. To the best of our knowledge there are reports of 15 cases of adrenoleukodystrophy presenting like spinocerebellar degeneration or olivopontocerebellar atrophy in the world literature. Recently, a novel point mutation (single nucleotide deletion in exon 2) of the ALD gene has been identified in patients presenting like spinocerebellar degeneration.[6] We could find out only two reports of adrenoleukodystrophy from India, presenting as adrenomyeloneuropathy.[7],[8] Extrapyramidal manifestations in adrenoleukodystrophy are distinctly rare. We could locate only one case of truncal dystonia as a part of the manifestation of severe childhood cerebral phenotype is reported in world literature.[9] CT and MRI findings have been reported in a similar case labeled ataxic variant of adrenoleukodystrophy.[10] CT revealed marked atrophy of the cerebellum and pons and bilateral low-density lesions in the deep while matter of the cerebellum. MRI showed these lesions more clearly, as well as other lesions in the middle and superior cerebellar peduncles, despite the absence of cerebral white matter involvement at the time of presentation. Management of these patients includes steroid replacement, Lorenzo′s oil, bone marrow transplantation and gene therapy. Lorenzo′s oil has been found to reduce the risk of developing MRI abnormalities in asymptomatic boys with normal MRI brain. Bone marrow transplantation is recommended for patients with mild but progressive neurological and MRI abnormalities in childhood cerebral phenotype. This case highlights the need for appropriate screening of all male patients with Addison′s disease for adrenoleukodystrophy and the need to keep high index of suspicion for the diagnosis of such patients because of its varied neurological manifestations. References
Copyright 2006 - Neurology India The following images related to this document are available:Photo images[ni06057f1.jpg] [ni06057f2.jpg] |
| |||||||||

{kind=link}
{kind=link}