 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
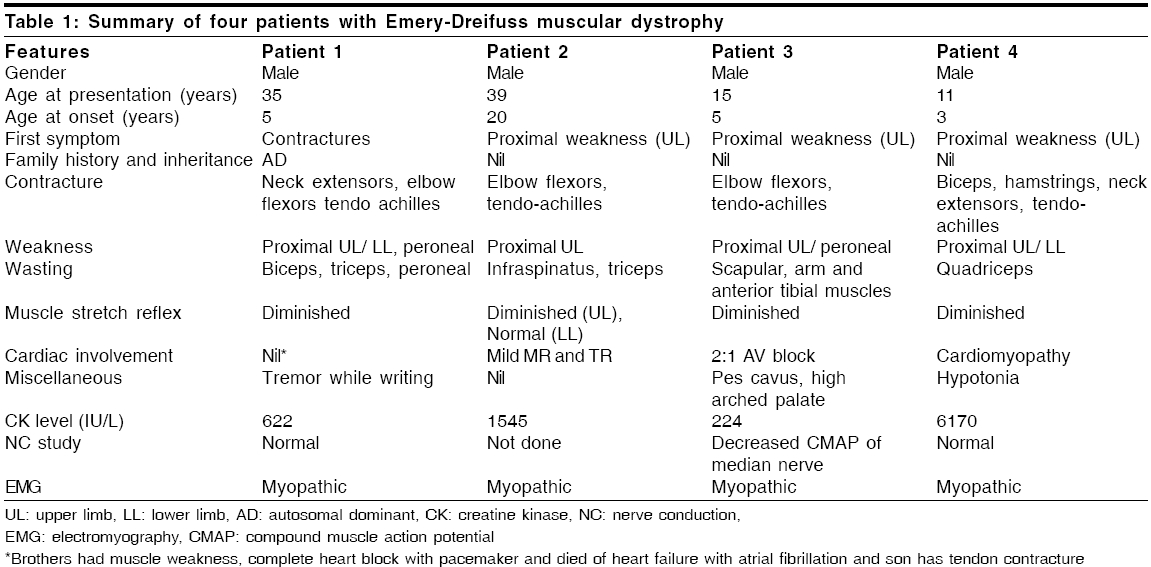
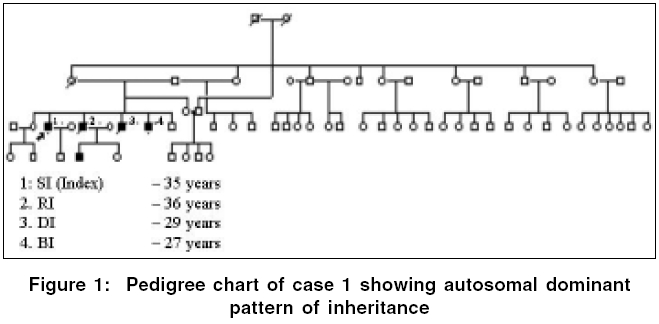
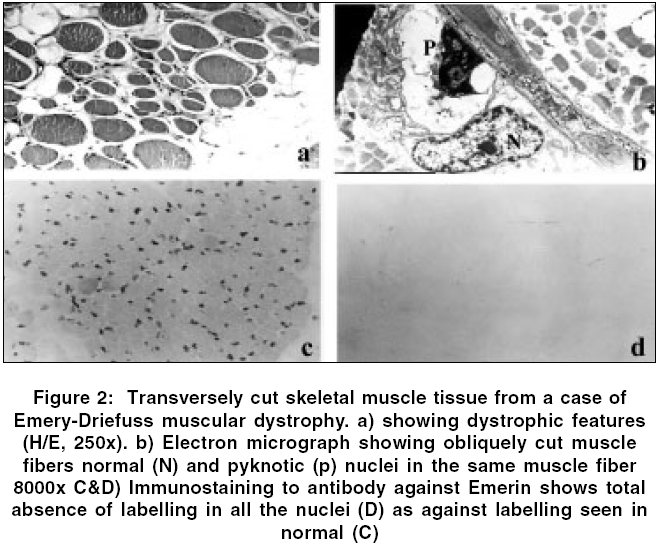
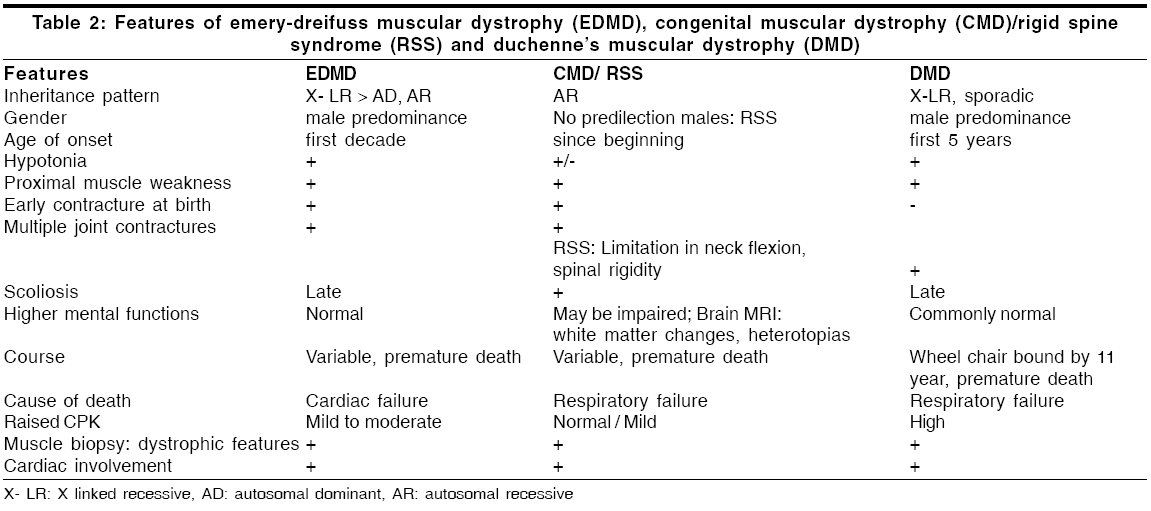
Neurology India, Vol. 54, No. 2, April-June, 2006, pp. 197-199 Case Report Emery dreifuss muscular dystrophy: A clinico-pathological study Gayathri N, Taly AB, Sinha S, Suresh TG, Gorai D Department of Neuropathology, National Institute of Mental Health and Neurosciences, Bangalore Code Number: ni06058 Abstract Emery-Dreifuss muscular dystrophy (EDMD) is a rare and genetically heterogeneous disorder. We report two patients with emerin deficient X-linked EDMD and two probable patients with EDMD with typical early contractures, progressive muscle weakness and cardiac involvement. Family history was noted in one case. Muscle biopsy revealed features of dystrophy in all.Keywords: Cardiac changes, contractures, Emery-Dreifuss muscular dystrophy Introduction Emery-Dreifuss Muscular Dystrophy (EDMD) clinically manifests with weakness and wasting of humero-peroneal muscles, early contractures of elbow flexors, tendo-achilles and paraspinal muscles and cardiac abnormalities.[1] Muscle biopsy shows dystrophic features. Diagnosis of X-linked recessive EDMD is established by absence of emerin or linking for mutation on STA gene. Mutations in Lamin A/C are seen in autosomal dominant (AD) more common and recessive (AR) forms,[2] however immunodiagnosis is not helpful as the protein is being continuously synthesized by the normal allele.[3] To the best of our knowledge there are no reports on EDMD from India. We report 2 patients with ′emerin deficient′and 2 probable patients with EDMD (lamin A/C deficiency). Materials and Methods Detailed phenotype was noted in all the patients. Laboratory evaluation included complete hemogram, urine analysis, serum biochemical analysis including muscle enzymes, ECG, 2D- ECHO and chest X-ray. Nerve conduction (NC) and electromyography (EMG) studies were performed. All patients underwent a quadriceps muscle biopsy. Serial sections were stained for light microscopy. Immunostaining to monoclonal antibodies against dystrophin, alpha-sarcoglycan, merosin and emerin as primary and HRP tagged goat anti-mouse as secondary was carried out in all except patient 1 (only paraffin sections available). For electron microscopy (EM), the specimen was fixed in 3% gluteraldehyde in phosphate buffer, post-fixed in 1% osmium tetroxide, dehydrated in graded series of alcohol and embedded in araldite. Thin sections contrasted with uranyl acetate and lead citrate were observed under EM. Results The case histories are summarized in [Table - 1]. All patients were male. Three patients had onset in the first decade. Weakness was progressive and involved proximal muscles initially. Wasting was variable but significant in weaker muscles. While all developed prominent contractures, patient 1 had noted it at onset. Cardiac involvement (2:1 AV block, valvular (Mitral Regurgitation, Tricuspid Regurgitation) pathology and cardiomyopathy) was observed in all except in-patient 1. Serum CK levels ranged from 1.5 to 36 times the normal value (170 IU/L). Patient -1 had AD inheritance [Figure - 1]. Two of his younger brothers had similar phenotype. One of them (DI, 29 years) had atrial fibrillation (AF), required pacemaker and died due to cardiac failure, while the other (BI, 27 years) who is alive, had supraventricular and ventricular ectopics. One of the elder brother (RI) had progressive limb weakness since 15 years. He had cardiac failure with AF requiring pacemaker and died at 36 years. Son of the index case developed contracture of tendo-achillis at 6 years of age. Routine histology revealed dystrophic features with increased collagen. Scattered atrophic fibers with clumped nuclei and type II fiber grouping and moth-eaten fibers were noted in patient 3 and 4 respectively. Immunostaining to antibodies against dystrophin, alpha-sarcoglycan and merosin showed normal localization. Antibodies against emerin revealed total absence of labeling in all the nuclei of the myofibres, fibroblasts and endothelial cells in-patients 2 and 3. Positive labeling was noticed in-patient 4. Immunostaining in-patient 1 was not carried out. EM revealed mild disorganization of myofilaments, displacement of mitochondria and proliferation of T-system in some of the fibers. Majority of the nuclei appeared normal. Some of the nuclei showed significant grooving and hyper- condensed chromatin. Similar changes were noticed in an occasional nucleus of fibroblasts and endothelial cells. These nuclear changes were observed in disorganized and normal appearing fibers. In longitudinal sections, altered and normal appearing nuclei were present in the same fiber [Figure - 2]. The nuclei under EM in probable EDMD (patient 4) were normal. Discussion Emery had described this entity with triad of humero-peroneal weakness, early contractures and cardiac disturbance.[4] Miller et al[5] proposed diagnostic criteria: (a) early contractures of Achilles-heel, elbows and spine; (b) slow progression and symmetric weakness prominent in humero-peroneal muscle; (c) cardiac conduction abnormality and/or cardiomyopathy; (d) myopathic features; and (e) X-linked inheritance. Contractures occur even before onset of weakness and are attributed to primary abnormality of the tissue that surrounds the joints or secondary to dystrophic changes in the muscle. Weakness often involves humero-peroneal muscles, however scapulo-pelvic and humero-pelvic distributions have also been reported.[6] Our patients had all these phenotype. There are reports of clinical overlap between EDMD, congenital muscular dystrophy/ Rigid Spine Syndrome and Duchenne′s muscular dystrophy (DMD).[7] [Table - 2] Padma et al[8] had reported 1 patient of DMD clinically misdiagnosed as EDMD. Cardiac involvement was noticed in cases 2, 3 and 4. One of the cases (case 2) was on pacemaker. Even though case 1 had no obvious cardiac involvement, there was strong family history of cardiac rhythm disturbances including sibling deaths due to cardiac failure. Intrafamilial variation among AD-EDMD is known. First-degree AV block and complete heart block are common followed by paroxysmal atrial tachycardia, AF, atrial flutter, bundle branch block, second-degree AV block and ventricular tachycardia. Cardiac symptoms appear between the 2nd and 5th decade of life and may result in sudden death.[9] Manilal et al (1999)[10] found absence of emerin and lamin in the nuclei of skeletal and cardiac muscle. Cardiac defects are attributed to deficiency of emerin and lamin. While Becker muscular dystrophy (BMD) and DMD arise from genetic defects in the dystrophin, EDMD arises from genetic defects in nuclear proteins (emerin and lamin A/C). X-linked, AD and AR inheritance EDMD has been described. The X-linked variant is caused by defects in the STA gene on Xq 28 (codes for 34 KD), a protein called emerin while the AD/AR is due to defects in the LMNA gene located at 1q21 that encodes lamin A and C.[6] There was AD pattern in-patient 1. Cases 2 and 3 in the present study showed total absence of emerin in all the nuclei confirming the diagnosis of X-linked Emerinopathy. Patient 4 had high CPK and showed positive labeling for emerin. A diagnosis of probable AD/AR EDMD was considered. Low penetrance of some mutation might explain this. EM examination revealed pyknotic nuclei in some of the fibers. Neither karyoplasm extrusion nor membrane bound channels, as described by Fidzianska et al ,[11] were observed. Emerin anchors the chromatin to the nuclear envelope.[12] The grooving and pyknosis of nuclei may be attributed to absence of emerin in the membrane. The combination of progressive muscle weakness and, in particular, the cardiac complications result in considerable morbidity and at times mortality. Early diagnosis might prevent cardiac death. Accurate identification of carrier status and prenatal prediction might help. Muscle biopsies may be avoided if immunostaining with emerin antibodies on oral exfoliates could be performed.[13] Immunohistochemistry is thus essential to categorize this disorder for genetic counseling. References
Copyright 2006 - Neurology India The following images related to this document are available:Photo images[ni06058t2.jpg] [ni06058f2.jpg] [ni06058t1.jpg] [ni06058f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}