 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
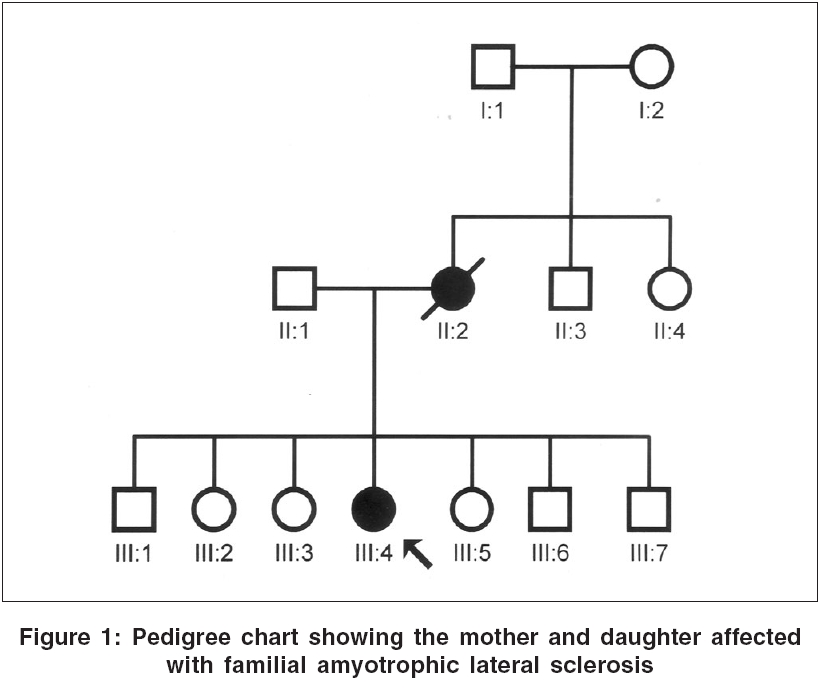
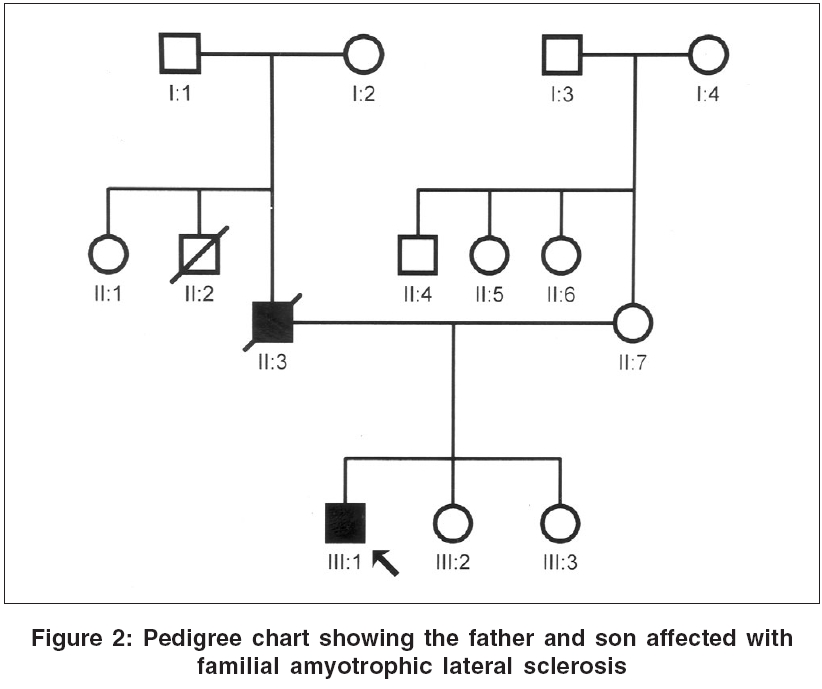
Neurology India, Vol. 54, No. 3, July-September, 2006, pp. 304-305 Case Report Familial amyotrophic lateral sclerosis: First report from India Nalini A, Yeshraj G, Veerendrakumar M Department of Neurology, National Institute of Mental Health and Neurosciences, Bangalore Date of Acceptance: 15-Feb-2006 Code Number: ni06098
We report two patients diagnosed to have familial amyotrophic lateral sclerosis (FALS). A 40 year old lady had progressive weakness and atrophy of the limbs and bulbar palsy from the age of 39 years and with electrophysiological evaluation was confirmed as definite ALS. Her mother had presented in 1978 at the age of 42 years with symptoms and signs of ALS. The other patient was a 43 year old male with rapidly progressive weakness, wasting and spasticity of the limbs and bulbar palsy of 4 months duration and with electrophysiological evidence of diffuse anterior horn cell involvement. His father also had onset of illness at 43 years of age with gradually progressive spasticity and atrophy of the extremities followed by bulbar palsy. In the first instance the mother had a duration of illness of 8 years while in the second the father lived for 15 years after the onset of illness. Keywords: Familial amyotrophic lateral sclerosis, India Introduction Amyotrophic lateral sclerosis (ALS) is commonly a sporadic disorder. In 5-10% of patients, there is a family history of similar illness, usually with an autosomal dominant inheritance.[1],[3] There is no report on Familial ALS (FALS) from India, although many are published from elsewhere. We report the clinical profile and investigation results of two patients with FALS seen at our institute.Case Reports Case 1 A 40-year-old lady presented in 2001 with progressive weakness and spasticity of lower limbs of 12 months duration. Six months later, she noticed weakness of both hands, followed by generalized fasciculations, dysarthria, dysphagia and had became dependent for most activities by the end of 9 months. Examination showed normal higher mental functions, mixed dysarthria, sluggish palatal reflex and wasting of the tongue. Motor system revealed fasciculations, severe wasting of hand and feet muscles and spasticity in all limbs. Power was MRC grade 2-4 in upper limbs and 3-4 in lower limbs. Tendon reflexes were exaggerated and plantar response was extensor. Jaw jerk was brisk. She could walk independently. Routine blood investigations, thyroid function tests, collagen vascular work-up, protein electrophoresis were normal. Malignancy work-up was negative. Electromyography in all limbs revealed active and chronic denervation with reinnervation. Nerve conductions were normal. The proband′s mother, aged 42 years, was seen at our institute in 1978 [Figure - 1]. The illness had begun with distal weakness and wasting in the left lower limb and left hand of 18 months duration. Two months later, she developed weakness and wasting of right hand, followed by involvement of distal muscles of right lower limb with fasciculations. By 12 months, she developed bulbar palsy and emotional incontinence; and by 14 months, she was wheel-chair-bound. Examination had showed normal higher mental functions, bulbar palsy, brisk jaw jerk, spasticity with fasciculations and atrophy of distal muscles of upper limbs with power of grade 3 in proximal and grade 2 in the distal groups. Tendon reflexes were exaggerated with extensor plantar response. Routine investigations, thyroid function tests, myelography and cerebrospinal fluid analysis were normal. She had lived for 8 years and then expired. Case 2 A 43-year-old male presented in 2003 with progressive spasticity, weakness and atrophy of right lower limb of 4 months, followed by similar symptoms in the other limbs with fasciculations. One month later, he developed bulbar palsy. He had severe mixed dysarthria, bifacial weakness, sluggish palatal reflex, wasting of the tongue, hypotonia in upper limbs, spasticity in lower limbs and severe atrophy of limb muscles with grade 2 to 3 power. Tendon reflexes were exaggerated with extensor plantar response. Serum electrophoresis, bone scan, MRI of brain and cervical spinal cord were normal. Electromyography revealed active denervation and chronic reinnervation in all the limbs. Nerve conductions were normal. The proband′s father, at 43 years of age, had developed difficulty in walking with stiffness of lower limbs [Figure - 2]. Over subsequent 3 years, he noted weakness and atrophy of upper and then lower extremities and required minimal support to walk; and in the 7th year, he developed progressive bulbar palsy. During the 10th year of illness, he became wheel-chair-bound. Fifteen years after the onset, he expired. Routine investigations, including imaging, were normal. Discussion Both of our index patients had presented with features suggestive of pure motor system involvement affecting upper and lower motor neurons, innervating limb and bulbar musculature. Electromyography and nerve conduction studies further confirmed the diagnosis of ALS in both of them. According to El Escorial criteria, both patients′ parents had clinical features of definite ALS. These observations confirm that both of our patients had FALS. Though ALS was described more than 100 years ago, FALS has only been recognized in the last five decades after the studies of Kurland and Mulder.[1] Most of the current literature suggests involvement of Copper-Zinc superoxide dismutase 1 (Cu-Zn SOD1) in the pathogenesis of FALS.[1],[2] So far, more than 50 missense Cu-Zn SOD1 mutations have been described.[4] Different SOD1 mutations cause distinct syndromes that differ with respect to penetrance, age of onset, survival and clinical manifestations.[5],[6] The onset of symptoms in the limbs and survival for 8-15 years without any ventilatory assistance in our patients further reiterates the diagnosis of FALS. Out of 850 patients with ALS seen at our institute over the last 40 years, these are the only two families with clinically confirmed FALS. Although there were a few other patients who reported that either their siblings or parent suffered from a similar illness, no documented evidence was available. These observations suggest that FALS is rather rare among Indian patients. Further research in this area warrants identification of more patients with FALS and their genetic analysis. References
Copyright 2006 - Neurology India The following images related to this document are available:Photo images[ni06098f2.jpg] [ni06098f1.jpg] |
| |||||||||

{kind=link}
{kind=link}